Drug Discovery - Yesterday and Tomorrow: The Common Approaches in Drug Design and Cancer
<p>The process of drug discovery has undergone radical changes and development over years. Traditionally, the drugs were discovered by employing chemistry and pharmacology-based cautious approach. When natural products were the most important source of drugs or drug precursors, but the conventional randomized drug research phenomenon was no longer effective at that time due to many negatives of these approaches like: high expenses of discovering new drugs, time-consuming and reduced success guarantee. Thus, with the development of the era, the concept of “Rational Drug Design†has enabled drug target identification and validation to be more specific. In addition, several novel technologies and approaches have been introducing economics, proteomics and other omics areas such as 3D QSAR, pharmacophore modeling and other, which playing a promising role in accelerating the pace of drug discovery process. Their view of the current research focuses on the importance of drug discovery in modern times and shows how old methods have been replaced and summarized. Some of examples of molecules are identified in addition to computational approaches used to discover it, specifically in the field of anticancer drug design</p>
Introduction
In 1945, the Nobel Prize in Physiology or Medicine was granted together to Sir Howard Walter Florey, Ernst Boris Chain and Alexander Fleming, the latter discovered penicillin from two decades before [1]. During the past 50 years, the goal of drug discovery has been to design compounds that interact with biological targets, according to a ‘one-drug–one-target’ paradigm [2]. Drug discovery process was a significant issue in the pharmaceutical industry because it was a very costly and time-consuming process to produce new drug potentials, although that was only a very limited number of drug discovery projects would lead to the discovery of a new drug [3, 4, 5]. Today, with the development of bio informatics techniques, it is Cell & Cellular Life Sciences Journal
possible to facilitate drug Discovery and product development at a faster rate by reduce the time, cost and make the people understand the complex mathematical and statistical equation in simple form. Bioinformatics is the branch that deals with the application of computer technology for the management and analysis of the biological information by making a combination among computers, databases, statistics, graphs and 3-d plots [6]. Many bioinformatics technologies are growing importance fields to understand and predict the potential drug because it focuses on two main areas: Data Management and Data Analysis [6]. The bioinformatics and computer-aided drug design (CADD) approaches play a critical role in addressing different challenges in drug design. It is the advance in understanding that a drug is an effective ligand for a protein of therapeutic interest in addition to the molecule need to have drug-like properties. Rational Drug Design (RDD) is basically a computer-aided molecular modeling which is a repetitive process that is based on the knowledge of three - dimensional (3-D) structure of the target proteins of interest. Such knowledge allows designing molecules capable of binding the receptor to maximize drug affinity and specificity towards the target. The major goal of the CADD center is to initiate these collaborations leading to the establishment of research projects to discover novel chemical entities with the potential to be developed into novel therapeutic agents. An advance in RDD is tightly coupled to advances in new algorithms for Computer – Assisted Molecular Modeling (CAMD). Computer Aided Drug Design (CADD) represents more recent applications of computers as tools in the drug design process. In most current applications of CADD, there are attempts to find a ligand that will interact favorably with a receptor that represents the target site [7]. With the dramatic increase of information available on genomics, small molecules, and protein structures, computational tools become integrated at almost every stage of the drug discovery and development. More recently, many cases of successful applications of structure-based drug design and Ligand -based drug design approaches have been reported. It is the case for designing potential anticancer drugs and drug candidates, the 3D structure of a target molecule, chemical compounds which may have a potentially higher affinity for their target when are designed rationally with the aid of computational methods [8]. Computational models generate useful predictions to be checked with experimental results, and biologists and physicians demand approaches that are more accurate to computational scientists [9]. In the current literature, the main objective of this research was to review the drug discovery in the past and to show how effect development of the computational drug design approaches on the design of new drugs in directing further therapy in future. Drug discovery is discussed further in the following section.
Drug Discovery
More than 30years ago, Macro molecular X-ray crystallography was an important and powerful technique used by pharmaceutical companies in drug discovery. The crystal structures of protein–ligand complexes allows the study of the specific interactions of a particular drug with its protein target at the atomic level and it is used to design and improve drugs discovery [10]. The utility of this technique is demonstrated in the discovery and optimization of a new orally available class of urokinase inhibitors for the treatment of cancer [11]. In the design of inhibitors of serine proteases to control blood clotting and in Fragment-based drug discovery which allows the complementarily between a protein active site and drug- like molecules to be rapidly and effectively explored, using structural methods but most pharmaceutical companies considered X-ray crystallography too expensive and time-consuming [12, 13, 14]. Traditionally, the typical drug discovery and development cycle, from concept to market is a very difficult task. It has many limitations such as: The drug discovery takes approximately 14 years because synthesizing compounds of drugs is a multi-step and time-consuming process. Therefore, the most successful pharmaceutical companies have only one project success from ten when they are bringing a drug to market. Such process has resulted in high attrition rates with failures attributed to poor pharmacokinetics (39%), lack of efficacy (30%), animal toxicity (11%), and adverse effects in humans (10%) [15, 16]. Drug design projects can fail because of the lack of the adequate assays or animal models to test for the proper functioning of candidate compounds. Another limitation from the drug-design limitations is that of the compounds that are active against the disease which may be too toxic, not bio available, or too costly to manufacture. There cent estimates of the costs to bring a drug to the market have ranged from $300 million to $1.7billion. A single laboratory researcher’s salary, benefits, laboratory equipment, chemicals, and supplies can cost in the range of $200,000 to $300,000 per year [17]. In addition to that, Cell & Cellular Life Sciences Journal
some diseases are so rare that the cost of a development effort would never be covered by product sales. Figure 1 refers to the traditional life cycle of drug discovery. Currently, the rapid developments in combinatorial chemistry and high – throughput screening technologies have provided an environment to expedite the drug discovery process by enabling huge libraries of compounds to be screened and synthesized in a short time. Although the investment in new drug development has grown significantly in the past decades, the output is not positively proportional to the investment because of the low efficiency and high failure rate in drug discovery [18]. Consequently, various approaches have been developed to shorten the research cycle and reduce the expense and risk of failure for drug discovery. The bioinformatics and Computer aided drug design (CADD) is one of the most effective methods for reaching these goals [19].
$$ \mathrm {S y n t h e t i c o r N a t u r a l C o m p o u n d} $$
$$ \mathrm {P r e c l i n i c T r a i l s} $$
Drug Design Approach
In the most basic sense, the drug design is the design of small molecules that are complementary in shape and charge to the bio molecular target to which they interact and bind [20]. Drug design is referred to as rational drug design (or more simply rational design) [21]. The objective of drug design is to find a chemical compound that can fit geometrically and chemically to a specific cavity on a protein target [22]. Traditional methods in the design of new drugs which discovered by coincidence or trial and error methods were replaced as a result of the development of new approaches and technologies that yield significant savings in time and money and increase in diversity and specificity of lead compounds. The development of in Silico or “Computer aided drug design (CADD)” has been receiving more and more attention in the worldwide [20, 21]. Current approach in drug discovery needs the understanding of the disease mechanism, pathways, identifying disease causative proteins followed by target identification and lead compound discovery [23]. Nowadays, predominant methods of drug design and discovery are integrating techniques of x-ray crystallography, computational chemistry, and nuclear magnetic resonance spectroscopy. Also, the research activity is being mimic of the Dear Dr. Li Pin Kao, complex molecular interactions of natural proteins by focusing on small-molecule structure-based drug design, In Silico drug design can be applied according to two approaches of drug design depending on the knowledge of the target, presence of the primary sequence, and 3D structure. These strategies are: Structure – Based Drug Design (SBDD) and Ligand – Based Drug Design (LBDD) [21, 24, 25]. Structural – Based Drug Design is generally the preferred method of drug design because it is the process that incorporates both experimental and computational techniques. In addition, it is not a single tool or technique of drug designs in ceit has the highest success rate and the docking, as well as in it is the preferred tool for giving a computational prediction of compound activity [26]. Figure 2 is an example of docking process.
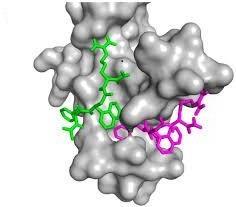
Structure Based Drug Design
In the drug design, one of the major challenges in computational approaches is the accurate prediction of Cell & Cellular Life Sciences Journal
the binding affinities between small molecules and their macro molecular receptors, SBDD approaches are responsible for that [28, 29]. Structure-based drug design (SBDD) methods are becoming increasingly powerful, versatile and more widely used [30]. If the target 3D structure is known, it can be use Structure-Based Drug Design (SBDD) strategy for the design of new ligands, which it is a well-established as successful and highly attractive strategy used by academic and pharmaceutical research laboratories worldwide [31, 32]. The identification of peptide-based HIV protease inhibitor was the first success of structure based drug design [33] and inhibitors against MDM2 protein which interact with P53 and cause cancer [34]. From the first published work describing SBDD in 1976 to the present day the computer aided molecular design and SBDD have played a key role in the development of several marketed drugs [35].
Ligand Based Drug Design
Ligand based drug design is one of the popular approaches for drug discovery and lead optimization used when the 3D structure of a target protein are unavailable, new drugs will designed based on the knowledge of already reported drugs for that particular biological target [36, 37]. From the successful example of small molecules designed using a ligand-based approach is the case of tubul in polymerization inhibitors, which is an essential component of cell cycle progression and cell division represents an important target for anticancer therapy [38]. Many methods are used in the ligand – based drug design process, but the 3D structure-activity relationships (3D QSAR) and pharmacophore modeling are widely used because they are considered as the most important tools. Which can provide predictive models which are suitable for lead compound optimization in addition to the crucial role in the nature of the interactions between drug target and ligand molecule? [39]. 3dqsar: Quantitative structure-activity relationships (QSAR) are statistically derived models from the knowledge of chemical structure it can predict the physicochemical and biological properties of molecules. Classical QSAR methods describe relationships using mathematical models which further validate and predict the model statistically. Predicting the biological activity of untested compounds from their molecular structures is one of the major applications of QSAR model. Overcoming the critical problem in QSAR modeling in estimating the accuracy of prediction becomes easy after the development of molecular modeling. Three-dimensional (3D) descriptors have replaced the traditional physicochemical and bi-dimensional descriptors [40]. A 3D-QSAR is a mathematical attempt to define the properties of the active site without knowing its structure. This is done by computing the electrostatic and steric interactions that an imaginary probe atom would have if it were placed at various positions on a grid surrounding a known active compound [17]. 3D QSAR is a well- established, successful and highly attractive strategy which allows the identification of the pharmacophoric arrangement of molecular fragments in space and provides guidelines for the design of the next generation of compounds with enhanced biological potencies. Therefore, it is used by academic and pharmaceutical research laboratories worldwide [31, 32, 41, 42]. Pharmacophore Modeling: Pharmacophore approaches have become one of the major and successful computational tools in drug discovery [43, 44]. The concept of pharmacophore was first introduced in1909 by Ehrlich [45]. Who defined the pharmacophore as the essential features responsible for a drug’s biological activity and it is the first essential step towards understanding the interaction between a receptor and a ligand [43, 46]. After the past century’s various ligand- based and structure-based methods have been developed for improved pharmacophore modeling and the latest can be established either by superposing a set of active molecules and extracting common chemical features that are essential for their bioactivity in a ligand-based manner, or by probing possible interaction points between the macromolecular target and ligands by in a structure-based manner. Pharmacophore approaches have been successfully and extensively applied in virtual screening, de novo design and other applications such as lead optimization and multi target drug design [44, 47, 48]. Although in recent years, many successful stories of pharmacophore approaches in facilitating drug discovery have been reported, still pharmacophore faces many challenges that limit its capability to reach its expected potential, particularly with reducing the high cost associated with the discovery and development of a new drug. However, with success stories in drug discovery and increasing application ranges of pharmacophore, enable further enrichment of the pharmacophore concept and development of it. [44]. Others: Many other methods used in the ligand–based drug design process such as: CoMFA and CoMSIA, the first one developed as a tool to study 3D QSAR that begins with a traditional pharmacophore modeling to suggest a Cell & Cellular Life Sciences Journal
bioactive conformation for each molecule and to superimpose the molecules under study. Although CoMSIA method based on similarity indices similar to CoMFA but it is overcomes to the problems associated with the functional form of the Lennard-Jones potentials used in most of the CoMFA methods because this method adopted Gaussian type functions instead of traditional CoMFA potentials [49, 50]. GRID and GOLPE is the other method used in the ligand – based drug design which is better than COMFA method because of reduced number of potential functions to (6-4) compared to Lennard-Jones potential (6-12) in COMFA [51]. In recent years, QSAR validation received more attention that there were four tools to validate a QSAR model
- Randomization of the response data
- Cross-validation
- Bootstrapping
- External validation by splitting the total data set into test and training set.
REACH (Registration, Evaluation and Authorization of Chemicals) legislation enforced in the European Unions agreed that QSAR models should be validated scientifically and regulatory bodies should take decisions based on sound scientific background [52].
| Compound name | Drug target | C | omputational approac | h | Therapeutic area | R | eferenc | e | ||||||
| Gefitinib combination with imatinib | allosteric site of Abl kinase | MTT cell proliferation assay | treatment of CML | [53] | ||||||||||
| ZINC08764498 (hit1) and ZINC12891610 (hit2) | Bcr-Abl protein | Glide software | treatment of CML | [54] | ||||||||||
| FDA approved drugs | cancer patient- specific protein network maps based on the patient's | Integrated genomics and Computational biology modeling (CBM) approach. | Patient’s with Relapsed/refractory ETP- ALL. | [55] | ||||||||||
| Drug target discovery | BCL2, caspase-3 and TP53. | Construction of a cancer- perturbed protein-protein interaction network | Potential molecular targets for development of anticancer drugs. | [56] | ||||||||||
| α-lipoic acid shikonin ester derivatives | against both mitosis (tubulin) and glycolysis (PDK) | computer assistant drug design method | clinical anticancer agent | [57] | ||||||||||
| Brk (also known as protein tyrosine kinase 6, PTK6) | 19,20-anhydrosipholenol A 4-β-benzoate | a kinase assay profiling platform | controlling breast cancer proliferation and migration | [58] | ||||||||||
| indazole-based diarylurea derivatives | targeting c-kit | Structure-based drug design. | colon cancer HCT-116 cell line and hepato cellular carcinoma PLC/PRF/5 cell line | [59] | ||||||||||
| 2,4-dihydroxy benzaldehyde | a molecular chaperone (Hsp90) | a validated molecular docking methodology | Anti proliferative effect against PC3carcinoma cells. | [60] | ||||||||||
| MI-888 | p53–MDM2 interaction | Spirotry prostatin A and alstonisine | anticancer agents | [61] | ||||||||||
| Danshensu | HRas | inverse docking (Pharm Mapper and id Target servers) | Anticancer target | [62] | ||||||||||
| carbonucleosides | Ras FTase inhibitors | A topological sub structural approach to molecular design (TOSS-MODE) | Anticancer Compounds | [63] |
Table 1: Selected inhibitors, drug targets and computational methods used for anticancer identification and interaction predictio
Computational models are mathematical models used to study the complex systems numerically by making a computer simulation and predictions of the system's behavior under different conditions [64]. It has become an important component of many drug discovery programs and provided fruitful insights into the field of cancer, because computational models given the advantage that much less investment in technology, resources, and time are required [65, 9, 8]. In recent years, many successful applications of the structure-based drug anticancer design have been reported. For example, identification of p53 unregulated modulator of apoptosis (PUMA) inhibitors; PUMA inhibition increased risks for cancer development and therapeutic resistance because it leads to apoptosis deficiency. This cancer-treatment target is central in mitochondria-mediated cell death by interacting with all known anti apoptotic Bcl-2 family members, such compounds have been identified through computational modeling, structure-based design, and high-throughput screening of natural product and synthetic libraries [8, 66, 67]. In this review, we summarize leading computational techniques of anticancer in Table 1 that include target prediction, compound name with Computational approach and therapeutic area. Currently, a rational drug design technique has become an indispensable instrument for the development of the target-based therapies [65]. Specially, in modern biology with development of medicine bio informatics which become essential for management of data and clinical applications of these data in drug design and development [68]. From the example of the application of bioinformatics in new therapeutic advances is the development of designer targeted drugs such as imatinib mesylate (Gleevec), in chronic myeloid leukemia which is interferes with the abnormal protein made [69]. Also, from the latest advances in bioinformatics tools which lead to discovering multi-targeting molecules, where a single chemical entity can act on multiple molecular targets. Nowadays, it is gaining a great importance in anticancer drug discovery. In this field, a promising computational approach for identification of target combinations, and virtual screening for the design of multi- targeting ligands include data mining, ligand and structure-based analyses [70]. Design I-Kappa-B KinaseB (IKK-B) inhibitor, is another example of design a small molecule using a computational approach which is a key player in the NF- B signaling pathway, represents yet another potential target for the treatment of cancer in addition to inflammation [71]. Researchers are also exploring peptides which are called ‘alternative scaffolds’ that enhance tumor penetration. With the development of these approaches new opportunity become available for impactful cancer treatments [72]. It is expected that rational poly pharmacology will play an increasingly important role in drug discovery during the near future.
Conclusion and Future Trends
This brief review found that the bioinformatic and the computer-aided drug design (CADD) center were created to foster collaborative research among biologists, biophysicists, structural biologists and computational scientists. Now a day, it is possible to design a drug using high technologies and which create a new area of drug design and development. As structural genomics, bioinformatics, and computational power continue to explode with new advances and further successes in structure-based drug design. New targets are being identified each year, structures of those targets are being determined at an amazing rate, and our capability to capture a quantitative picture of the interactions between macromolecules and ligands is accelerating [73]. For example, after the completion of human genome project, pharmacogenomics becomes the new area of research which evaluates the effect of genes and their polymorphism on drug response and makes a revolution in the drug discovery, development process and formulation optimization based on the evaluation different genetic markers. Furthermore, medicine will be smarter, safer and more efficacious based on pharmacogenomics approaches [74]. Another example is that deep learning methods will become a major computer-aided drug design (CADD) approach in the near future. Despite the advantages and popularity of using machine-learning approaches (e.g., QSAR) in modeling studies, machine intelligence has been replaced by the deep learning in recent years because it can deal with complex tasks based on large, heterogeneous, and high-dimensional data sets without the need for human input. These methods have been shown to be useful in drug design studies [75]. Also, from a new paradigm in drug discovery is of the Poly pharmacology, which is the process off in ding new uses for existing approved drugs which focuses on multi-target drugs (MTDs), has potential application for drug repurposing, prediction of off-target toxicities and rational design of MTDs. The computational strategies have important role in it [76].
Cell & Cellular Life Sciences Journal
In future, many novel technologies and methodologies will be developed to increase the efficiency of the drug discovery process, and many drug discovery programs have depended on these computational methodologies, from hit identification to lead optimization. However, computational models generate useful predictions to be checked with experimental results. We hoped to improve of the success rate of new drugs in the clinic and the finding of new uses for existing drugs by achieving a systems-level understanding of human diseases. Although the brief review of drug design was not completely accurate, it can serve as a foundation that covers some studies in this field to which improvements can be made in the future.
References
-
Arriaga EE (1968) New life tables for Latin American populations in the nineteenth and twentieth century’s. New life tables for Latin American populations in the nineteenth and twentieth century’s. 3: 324.
-
Hopkins AL (2008) Net work pharmacology: the next paradigm in drug discovery. Nature chemical biology 4(11): 682-690.
-
Rao VS, K Srinivas (2011) Modern drug discovery process: an insilico approach.Journal of Bioinformatics and Sequence Analysis 3(5): 89-94.
-
Arrowsmith J, Miller P (2013) Trial watch: phase II and phase III attrition rates 2011-2012. Nature Reviews Drug Discovery 12(8): 569-569.
-
Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, et al. (2015) Ananalys is of the attrition of drug candidates from four major pharmaceutical companies. Nature reviews Drug discovery 14(7): 475-486.
-
Shah VA , Rathod DN, Basuri T, Modi VS, Parmar IJ, et al. (2015) Applications of bioinformatics in pharmaceutical product designing: areview. World Journal of Pharmacy and Phar‐maceutical Sciences 4: 477-493.
-
Siju EG, Rajalakshmi R, Paulose AP, Fathima T, Dhanya PP, et al. (2017) Cadd: Pharmacological Approaches in Drug Design and drug Discovery 6(4): 892-908.
-
Mustata G, Li M, Zevola N, Bakan A, Zhang L, et al. (2011) Development of small-molecule PUMA inhibitors for matting radiation-induced cell death. Curr Top Med Chem 11(3): 281-290.
-
Prada-Gracia D , Huerta-Yépez S, Moreno-Vargas LM (2016) Application of computational methods for anti cancer drug discovery, design, and optimization .Boletín Médico Del Hospital Infantil de México(English Edition) 73(6): 411-423.
-
Carvalho AL, Trincão J, Romao MJ (2009) X-ray crystallography in drug discovery. Ligand-macro molecular interactions in drug discovery: methods and protocols 572: 31-56.
-
Nienaber VL, Richardson PL, Klighofer V, Bouska JJ, Giranda VL, et al. (2000) Discovering novelligands for macro molecules using X-ray crystallographic screening. Nature biotechnology 18(10): 1105-1108.
-
Beddell C, Goodford PJ, Norrington FE, Wilkinson S, Wootton R (1976) Compounds designed to fit a site of known structure in human haemoglobin. Br J Pharmacol 57(2): 201-209.
-
Davies TG, Tickle IJ (2012) Fragment screening using X-ray crystallography, in Fragment-Based Drug Discovery and X-Ray Crystallography. Top Curr Chem 317: 33-59.
-
Jhoti H, Cleasby A, Verdonk M, Williams G (2007) Fragment-based screening using X-ray crystallography and NMR spectroscopy. Curr Opin Chem Biol 11(5): 485-493.
-
Myers S, Baker A (2001) Drug discovery—an operating model for a new era. Nat Biotechnol 19(8): 727-730.
-
Kalyani G, Sharma D, Vaishnav Y, Deshmukh VS (2013) A review on drug designing, methods, its applications and prospects. Int J Pharm Res Dev 5: 15- 30.
-
Young DC (2009) Computational drug design: a guide for computational and medicinal chemists: John Wiley & Sons.
-
Shekhar C (2008) Insilico pharmacology: computer- aided methods could transform drug development. Chemistry & biology 15(5): 413-414. Cell & Cellular Life Sciences Journal
-
Ou-Yang S, Lu JY, Kong XQ, Liang ZJ, Luo C, et al. (2012) Computational drug discovery. Act a Pharmacologic a Sinica 33(9): 1131-1140.
-
Seifert M, Kraus J, Kramer B (2007) Virtualhigh- through put screening of molecular data bases. Curr Opin Drug Discov Devel 10(3): 298-307.
-
Kleinberg M L, Wanke LA (1995) New approaches and technologies in drug design and discovery. Am J Health Syst Pharm 52(12): 1323-1336.
-
Kalita JM, Saha A, Nath D, Patangia U (2015) Advances in Computer Aided Drug Design. Universal J of Pharm Sci Res 1: 17-22.
-
Barabási AL, Gulbahce N, Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nature reviews genetics 12(1): 56-68.
-
Lee YV, Wahab HA, Choong YS (2015) Potential inhibitors for isocitrate lyase of Mycobacterium tuberculosis and non-M.tuberculosis: A summary. Bio Med research international 2015(2015).
-
Kapetanovic I (2008) Computer-aided drug discovery and development (CADDD): In silico- chemico-biological approach. Chemico-biological interactions 171(2): 165-176.
-
Ho LY, Liu P, Wang CM, Wu JJ (2008) The development of a drug discovery virtual screening application on Taiwan unigrid. Lecture Notes in Computer Science 5036: 38-47.
-
Sable R, Jois S (2015) Surfing the protein-protein interaction surface using docking methods: application to the design of PPI inhibitors. Molecules 20(6): 11569-11603.
-
Sippl W (2002) Development of biologically active compounds by combining 3DQSAR and structure- based design methods. Journal of computer-aided molecular design 16(11): 825-830.
-
McLellan JS, Chen M, Joyce MG, Sastry M, Stewart- Jones GB, et al. (2013) Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342(6158): 592-598.
-
Andricopulo AD, Salum LB, Abraham DJ (2009) Structure-based drug design strategies in medicinal chemistry. Curr Top Med Chem 9(9): 771-790.
-
Cavasotto CN, Orry AJ (2007) Ligand docking and structure-based virtual screening in drug discovery. Current topics in medicinal chemistry 7(10): 1006- 1014.
-
Cardoso CL, et al. (2006) Development and characterization of animmo bilize denzymereactor (IMER) based on human glycerol dehyde-3- phosphate dehydrogenase for on-lineen zymatic studies. Journal of Chromatography 1120(1-2): 151- 157.
-
De Clercq E (2009) The history of antiretrovirals: key discoveries over the past 25 years. Reviews in medical virology 19(5): 287-299.
-
Shangary S, Wang S (2009) Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol 49: 223-241.
-
Lounnas V, Ritschel T, Kelder J, McGuire R, Bywater RP, et al. (2013) Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput Struct Biotechnol J 5: e201302011.
-
Selassie C, Verma RP (2003) History of quantitative structure–activity relationships. Burger's Medicinal Chemistry and Drug Discovery.
-
Tintori C, Manetti F, Botta M (2010) Pharmacophoric models and 3DQSAR studies of the adenosine receptor ligands. Current topics in medicinal chemistry 10(10): 1019-1035.
-
Chiang YK, Kuo CC, Wu YS, Chen CT, Coumar MS, et al. (2009) Generation of ligand-based pharmacophore model and virtual screening for identification of novel tubulin inhibitors with potent anticancer activity. J Med Chem 52(14): 4221-4233.
-
Acharya C, Coop A, Polli JE, Mackerell AD Jr (2011) Recent advances in ligand-based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Current computer-aided drug design 7(1): 10-22.
-
Kubinyi H (1997) QSAR And 3DQSARin drug design Part 1: methodology. Drug discovery today 2(11): 457-467.
-
Jorgensen WL (2004) The many roles of computation in drug discovery. Science 303(5665): 1813-1818. Cell & Cellular Life Sciences Journal
-
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3(11): 935-949.
-
Güner O, Clement O, Kurogi Y (2001) Pharmacophore modeling and three-dimensional database searching for drug design using catalyst. Curr Med Chem 8(9): 1035-1055.
-
Yang SY (2010) Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug discovery today 15(11-12): 444-450.
-
Ehrlich P (1909) Über den jetzigen Stand der Chemotherapie. European Journal of inorganic Chemistry 42(1): 17-47.
-
Wermuth C, Ganellin R, Lindberg P, Mitscher LA (1998) Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure and Applied Chemistry 70(5): 1129-1143.
-
Kubinyi H (2006) Success stories of computer-aided design. Computer applications in pharmaceutical research and development 2: 377.
-
Mustata G, Follis VA, Hammoudeh DI, Metallo SJ, Wang H, et al. (2009) Discovery of Novel Myc−Max Heterodimer Disruptors with a Three-Dimensional Pharmacophore Model. Journal of medicinal chemistry 52(5): 1247-1250.
-
Cramer RD, Patterson DE, Bunce JD (1989) Recent advances in comparative molecular field analys is (CoMFA). Prog Clin Biol Res 291: 161-165.
-
Klebe G, Abraham U, Mietzner T (1994) Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem 37(24): 4130-4146.
-
Ragno R, Simeoni S, Valente S, Massa S, Mai A, et al. (2006) 3-DQSAR studies on histone deacetylase inhibitors. A GOLPE/GRID approach on different series of compounds. J Chem Inf Mode 46(3): 1420- 1430.
-
Pratim Roy P, Paul S, Mitra I, Roy K (2009) On two novel parameters for validation of predictive QSAR models. Molecules 14(5): 1660-1701.
-
Singh VK, Chang HH, Kuo CC, Shiao HY, Hsieh HP, et al. (2017) Drug repurposing for chronicmyeloid leukemia: insilico and invitro investigation of Drug Bank data basefor allosteric Bcr-Abl inhibitors. Journal of Biomolecular Structure and Dynamics 35(8): 1833-1848.
-
Parcha P, Sarvagalla S, Madhuri B, Pajaniradje S, Baskaran V, et al. (2017) Identification of natural inhibitors of Bcr‐Abl for the treatment of chronic myeloid leukemia. Chem Biol Drug Des 90(4): 596- 608
-
Turcotte M, Bosse RC, Hawkins KE, Meacham A, Vijay V, et al. (2017) Personalized Therapy Design for Early T-Cell Precursor Acute Lymphoblastic Leukemia (ETP-ALL) Using Computational Biology Modeling within Vitro Validation. Am Soc Hematology 130(s1): 1448.
-
Chu LH, Chen BS (2008) Construction of a cancer- perturbed protein-protein interaction net work for discovery of apoptosis drug targets. BMC syst boil 2(1): 56.
-
Lin HY, Han HW, Sun WX, Yang YS, Tang CY, et al. (2018) Design and characterization of αlipoicacylshikoninester twin drugs as tubulin and PDK1 dual inhibitors. Eur j med chem 144: 137-150.
-
Foudah AI, Sallam AA, El Sayed KA (2015) Discovery and Computer-Aided Drug Design Studies of the Anticancer Marine Triterpene Sipholanes as Novel P- gpand Brk Modulators, in Handbook of Anticancer Drugs from Marine Origin Springer pp: 547-569.
-
Chu YY, Cheng HJ, Tian ZH, Zhao JC, Li G, et al. (2017) Rational drug design of in dazole‐based diarylurea derivatives as anticancer agents. Chem Biol Drug Des 90(4): 609-617.
-
Gupta SD, Revathi B, Mazaira GI, Galigniana MD, Subrahmanyam CV, et al. (2015) 2,4- dihydroxylbenzal dehy dederived Schiff bases as small molecule Hsp90 inhibitors: Rational identification of a new anticancer lead. Bioorg chem 59: 97-105.
-
Yu BD, Yu Q, Liu HM (2015) Spirooxindoles: Promisings caffolds for anticancer agents. European journal of medicinal chemistry 97: 673-698.
-
Chen SJ, Ren JL (2014) Identification of a potential anticancer target of danshensu by inverse docking Asian Pacific Journal of Cancer Prevention 15(1): 111- 116. Cell & Cellular Life Sciences Journal
-
Estrada E, Uriarte E, Montero A, Teijeira M, Santana L, et al. (2000) A novel approach for the virtual screening and rational design of anticancer compounds. Journal of medicinal chemistry 43(10): 1975-1985.
-
Humphreys P (1990) Computer simulations in PSA: Proceedings of the biennial meeting of the philosophy of science association. Philosophy of Science 1990(2).
-
Katsila T, Spyroulias GA, Patrinos GP, Matsoukas MT (2016) Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J 14: 177-184.
-
Yu J, Zhang L (2009) PUMA, a potent killer with or without p53. Oncogene 27(S1): 71-83.
-
Zhang L, Ming L, Yu J (2007) BH3 mimetics to improve cancer therapy; mechanisms and examples. Drug Resist Updat 10(6): 207-217.
-
Bayat A (2002) Science, medicine, and the future: Bioinformatics. BMJ: British Medical Journal 324(7344): 1018-1022.
-
Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, et al. (2001) Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. New England Journal of Medicine 344(14): 1038-1042.
-
Raghavendra NM, Divya P, Sundeep K, Akhila M, Prasad SVUM (2017) Dual or multi-targeting inhibitors: The next generation anti cancer agents. European journal of medicinal chemistry 143: 1277- 1300.
-
Noha SM, Atanasov AG, Schuster D, Markt P, Fakhrudin N, et al. (2011) Discovery of a novel IKK-ß inhibitor by ligand-based virtual screening techniques. Bioorg med chem lett 21(1): 577-583.
-
Curtin CM, Castaño IM, O'brien FJ (2017) Scaffold‐Based micro RNA Therapies in Regenerative Medicine and Cancer. Advanced health care materials 7(1): 1700695.
-
Anderson AC (2003) The process of structure-based drug design. Chem boil 10(9): 787-797.
-
Gupta S, Jhawat V (2017) Quality by design (QbD) approach of pharmacogenomics in drug designing and formulation development for optimization of drug delivery systems. J Control Release 245: 15-26.
-
Lavecchia A, Cerchia C (2016) Insilico methods to address poly pharmacology: current status, applications and future perspectives. Drug discov today 21(2): 288-298.
-
Zhang, L, Tan J, Han D, Zhu H (2017) From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug discov today 22(11): 1680-1685.
- The Muculent Bleb-Mucinous Cystic Neoplasm-Hepatobiliary Region
- Insulin Sensitizers as Anti-Aging Agents: Unveiling Synergies with Albumin, GLP-1RA, Klotho Protein, and Metformin in the Quest to Combat Aging
- Reprogramming of GLP-1 Response at Prediabetes for the Prevention of Type 2 Diabetes: The Role of Albumin and GLP-1 Receptor Agonists
- The Mingled Allies-Combined Hepatocellular Carcinoma and Cholangiocarcinoma
- Compilation and Embodiment-Leydig Cell Tumour Testis
- Glucolipotoxicity: A Novel Different Perspective on the Causes of Cancer