Management of Hepatic Burkitt Lymphoma in Children
Hepatic Burkitt lymphoma is extremely rare in childhood and can be overlooked in differential diagnosis of liver masses. Patient: A8-year-old girl presented with a 1 month history of abdominal pain and weight loss and jaundice. Results: Physical examination revealed hepatomegaly and no palpable lymph node. Laboratory finding showed mild anemia (hemoglobin, 10,8 g/dL), elevated transaminase (ALT, 305 IU/L; ASAT,755 IU/L), elevated bilitubin (Bilirubin total, 179mg/L, Bilirubin direct, 143mg/l). Abdominal ultrasound shouwed a multiple hepatic lesions. Liver biopsy examination confirmed Burkitt's lymphoma. No metastasis was detected in the thoracic cavity, bone marrow, and spinal fluid. The patient was treated with the combination regimen of cyclophosphamide, doxorubicin, vincristine, prednisone and high dose methotrexate. Cytosine arabinoside and methotrexate were added for CNS prophylaxis by intrathecal installation. Serial follow-up ultrasound showed a marked decrease in the size of hepatic lesions but residual hilar lymph nodes at 2cm and the control showed stable size of lymph node after 28 months of chemotherapy. Conclusion: The clinical feature of primary hepaticlymphoma varies from no symptom to fulminant hepatic failure. There are no specific imaging criteria for diagnosing primary hepatic Burkitt’s lymphoma. Thus, histology by biopsy is necessary for diagnosis.
Introduction
Burkitt lymphoma (BL) is a highly aggressive B-cell non-Hodgkin lymphoma. Its primary hepatic’s localization is rare entity (PH-BL) [1]. We describe a case of PH-BL presenting with a jaundice and abdominal pain.
An 8 years old girl, presented with abdominal pain and jaundice and weight loss for 1 month before her admission. There was also a 3 days of history of moelena without fever or diarrhea. Physical examination revealed Gastroenterology & Hepatology International Journal
hepatomegaly measured 26cm below the subcostal margin, no palpable lymphnode or spleen. Laboratory findings were Hemoglobin 10,8gr/dL, Platelet 406000/mm3, WBC 13460/ mm3, alanin aminotransferase 3005(ALT) 23 IU/L, aspartate aminotransferase (AST) 755 IU/L, total bilirubin 179,3mg/L, direct bilirubin 143 mg/L and lactate dehydrogenase (LDH) 500/L. Alpha foetoprotein was normal at 8,9UI/ml. Coagulopathy test (Prothrombin time and partial thromboplastin time) was normal. Abdominal ultra sound showed multiple large low- density mass lesions in both lobes of liver with no enhancement, with hilary lymphnodes but no thrombosis was evident. No bile duct dilatation was noted (Figure 1). Chest x-ray was normal.
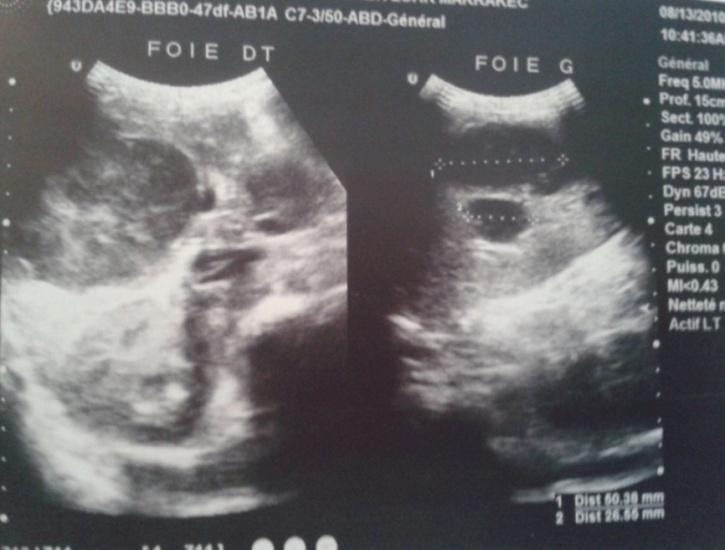
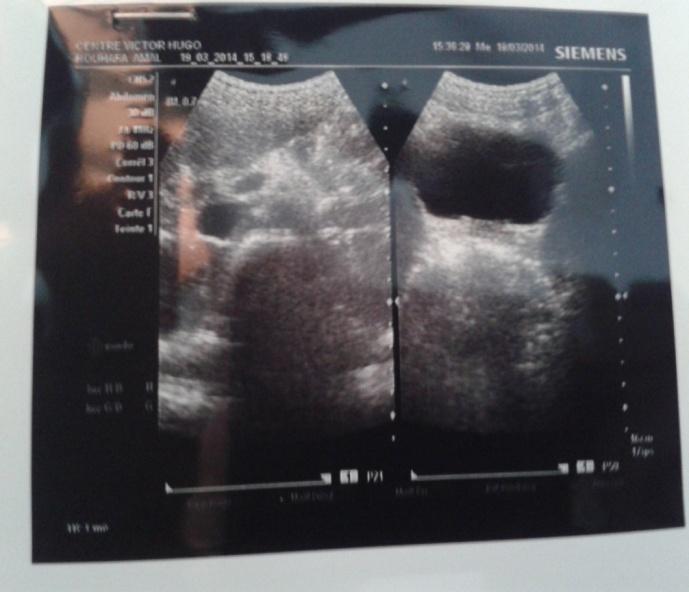
Figure 1: Chest x-ray. Ultra sonography- guided liver biopsy was done. Microscopic evaluation showed diffuse infiltration of monotonous small to medium sized cells with vesicular nuclei, dense chromatin pattern, prominent nucleoli. Mitotic figures were frequent, and many scattered tangible macrophages creating starry-sky pattern. Immuno histochemical study was not available. Spinal fluid analysis and bone marrow biopsy results were negative. Endoscopy, to explore hermeloena, was not done because patient didn’t present any episode at her admission. She received chemotherapy according to MAT IV (MoroccoAlgeria and Tunisia)/ GFAOP (Franco-africain pediatriconcology group) protocol. The patient had full regression of jundice at first and hepatomegaly since the first course. Her ALAT and ASAT levels normalized. Abdominal US control showed a regression of hepaticlesions and lymphnodes, but aprearance of coeliomesenteric adenopathy at 22mm which persisted since 28 months after the end of treatment and disapear after. Last control of patient were at 4 years after treatment and abdominal US was normal (Figure 2).
Discussion
According to Lei criteria in primary hepatic lymphoma symptom expression mainly originate from liver infiltration with no distant lymphadenopathy or leukemoid reaction in peripheral blood smear [2, 3]. PH- BL, a highly aggressive subset of NHL, is a very rare entity [2]. There have been no more than 15 cases of primary hepatic Burkitt’s lymphoma reported. Most of the reported patients were young, aged about 32 years old on average, and male, with a large proportion being children [4]. It is accounting for only 0.4% of all extra nodal lymphomas and make up to 3% of primary hepatic lymphomas [3]. It was a first case in our unit in Morocco. On histopathologic examination, Burkitt’s lymphomais defined as infiltration of neoplastic cells, which are marked lyuniform in size and shape. The nuclei are approximately the same size as the nuclei of the admixed Gastroenterology & Hepatology International Journal
histiocytes and therefore are smaller than the centroblasts and contain two to four basophilic nucleoli. The nuclear contours are generally round without deep indentation. The cytoplasmis strongly basophilic with small round cytoplasmic vacuoles best observed in air- dried touch imprints [5]. The presence of tangible body macrophages, phagocytosing abundant a poptotic debris creating starry- sky appearance is characteristic finding [6]. On immuno histochemical examination the neoplastic cells are positive for surface IgM and Ig light chain (κ>λ), pan –B- cell antigens (CD19, CD20, CD22, CD79α) and the germinal center associated markers such as CD10 and BCL6. The clinical feature of primary hepaticlymphoma varies from no symptom to fulminant hepatic failure. Fever, weightloss, night sweating (known as B symptoms), right upper quadrant pain, hepatomegaly, fatigue, jaundice, nausea, vomiting, and splenomegaly are common symptoms and rarely, bleeding tendency, ascites, pleural effusion, hepatic encephalopathy can occur [2]. But the common clinical manifestations in the reported cases were abdominal pain, pyrexia and the finding of hepatomegaly on physical examination [3]. There are no specific imaging criteria for diagnosing of PH-BL socytological or histological exams are necessary [3]. Early administration of multiple chemotherapeutic agents increases the curative potential of Burkitt’s lymphoma and overall survival make up 87% at 5 years after oncological treatment [2, 7]. Prognosis of patients with primary hepatic Burkitt’s lymphoma does not necessarily seem to be unfavorable [4]. Very high cure rates and long term survial are possible with intensive chemotherapy regimens [8].
Conclusion
Primary Hepatic Burkitt’s Lymphoma in an Immunocompetent Patient: Case Report and Review of the Literature, Open Journal of Blood Diseases 3(1): 53-56.
The best method for the diagnosis of hepatic lymphomais needle biopsy of the liver guided by radiological exam. Surgery in the management of PHBL is not well defined but aggressive combination chemotherapy is necessary.
References
-
Mattar WE, Alex BK, Sherker AH (2010) Primary hepatic Burkitt lymphoma presenting with acute liver failure J Gastro canc 41(4): 261-263.
-
Saffar H, Zamani H, Sotoudehmanesh R, Hashemi Taheri AP, Sotoudeh M, et al. (2011) Primary Hepatic Burkitt’s Lymphoma in a Patient With Acquired Immuno deficiency Syndrome, Iranian Journal of Pathology 6(2) : 97-19070.
-
Singh S, Rana Kundu P, Tanwar P, Chhabra S (2013) Primary hepatic Burkitts’s lymphoma in an immunocompromised adult, Clin Cancer Investigation Journal 2(3): 253-255.
-
Sekiguchi Y, Yoshikawa H, Shimada A, Imai H, Wakabayashi M (2013) Primary Hepatic Circumscribed Burkitt’s Lymphoma that Developed after Acute Hepatitis B : Report of a Case with a Review of the Literature, J Clin ExpHematop 53(2): 167-173.
-
Ioachim H, Medeiros L (2008) Ioachime’s Lymph Node Pathology. 4th (edn.), New York: Wiliams& Wilkins.
-
Lee SH, Kim HJ, Mun JS, Oh HC, Lee HW, et al. (2008) A case of primary hepatic Burkitt’s lymphoma. Korean J Gastroenterol 51(4): 259-264.
-
Page RD, Romaguera JE, Osborne B, Medeiros LJ, Rodriguez J, et al. (2001) Primary hepatic lymphoma: favorable outcome after combination chemotherapy. Cancer 92(8): 2023-2029.
-
Paydas S, Demiryurek H, Ergin M, Acikalin A (2013)
- Management of Gallbladder Perforations: A Review
- From The Mouth to the Gut: The Oral Microbiome's Role in Promoting Gastrointestinal Disease
- Case Report: Intraductal Papillary Mucinous Neoplasm (IPMN) Complicated by Portal Vein Plaquing and Biliary Obstruction Mimicking Pancreatic Metastatic Malignancy
- Management of Non-Cirrhotic Portal Hypertension during Pregnancy: A Review
- Effectiveness of Omeprazole versus Pantoprazole for Symptomatic Relief of Gastro-Esophageal Reflux Disease (GERD)/ Acid Peptic Disease (APD): A Real-World Evidence (RWE) Study
- Case of Splenic Infarction; A Rare Presentation of Complicated Enteric Fever in a Pediatric Patient