Adjunctive and Transformed Immunity: Histiocytic & Dendritic Cell Neoplasm
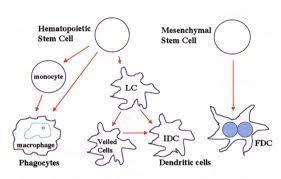
Exceptional malignancies of the lymph nodes or soft tissue comprising of < 1% of the tumour incidence are the Histiocytic and Dendritic cell neoplasm. A definitive appearance/biology/ Hematology/ histopathology / exclusive therapeutic options describe the condition. Morphology and Immune reactive appraisal may be mandated to distinguish the neoplasm. Preface: Histiocytic and Dendritic neoplasms are infrequent disorders which incriminate the accessory immune system or mesenchymal cells. Subject to the origin of the neoplasm, from the bone marrow precursors or the mesenchye, the lesions may be categorized as. i) The Histiocytic sarcoma (HS), Langerhans cell histiocytosis(LCH) and the Inter-digitizing dendritic cell sarcoma (IDCS) may commence from the bone marrow precursors and ii) The Follicular dendritic cell sarcoma (FDCS), Intermediate dendritic cell sarcoma (INDCS), Fibroblastic reticular cell tumour (FRCT) and Disseminated juvenile xanthogranuloma(DJX) may emanate from the Stromal or the mesenchymal cell. Divergent differentiation of the bone marrow precursors is a usual manifestation, though hybrid or trans-differentiating malignant lymphoid clones may also materialize in Inter-digitizing dendritic cell sarcoma (IDCS), Histolytic sarcoma (HS) and Follicular dendritic cell sarcoma (FDCS). Together, Histolytic and Dendritic cell tumors constitute < 1% of the lymph node or soft tissue neoplasm. The tumors may be misinterpreted as Non Hodgkin’s Lymphoma or adjunct Lymph-Proliferative disorders. The rare malignancies may be a challenge to discern and medicate. Debatable Histolytic and Dendritic neoplasm mandate a referral. The disorder may be ascertained by the combined analysis on Histology and Immune- histochemistry.
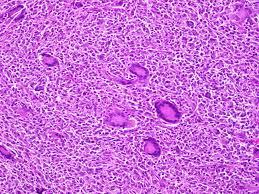
Follicular Dendritic Cell Sarcoma: “Morphology”
FDCS is an unusual neoplasm of the clone of follicular dendritic cells (FDCs) Follicular dendritic cells originate from the stroma [1, 2, 3], are usually located in the nodal germinal centers or in the extra-nodal ectopic lymphoid tissue such as the lymphoid aggregates of the bone marrow [3, 4, 5]. Immune complexes configured by the follicular dendritic cells routinely incorporate and accumulate antigens which provide a nidus for the propagation and identification of B cells, assisted by the T cells [6]. Follicular dendritic cells commence from the mesenchyme and may be identical to myo-fibroblasts. The cells may elucidate antigens akin to the bone marrow stroma, even though they do not arise from the bone marrow progenitors. Thus the immune markers pertaining to the follicular dendritic cell (FDC) differentiation may be elucidated such as CD21, CD23 and CD 35 [3]. Follicular lymphoma may probably be analogous to the mesenchyme generated Follicular dendritic cell sarcoma (FDCS) by the way of the trans- differentiation of the follicular lymphoma clone [1]. The sarcoma may be affiliated with Cattleman’s disease, Paraneoplastic Pemphigus and Myasthenia gravis [1, 2]. Follicular dendritic cell sarcoma (FDCS) may emerge in the lymph node which lodges the dysplastic follicular dendritic cells (FDCs) in the Cattleman’s disease. Follicular dendritic cell sarcoma( FDCS) due to the non- malignant follicular dendritic cells ( FDCs) of the Cattleman’s disease may expound Epidermal Growth Factor Receptor (EGFR) which may augment the follicular dendritic cells (FDC) perseverance with mutations that propound the Follicular dendritic cell sarcoma (FDCS)(1). Epstein Barr Virus (EBV) may be associated with the Follicular dendritic cell sarcoma (FDCS) [1, 2]. As the follicular dendritic cells (FDCs) demonstrate the CD21 molecule (operates as an EBV receptor), the virus may infiltrate the cells. Follicular dendritic cell sarcoma (FDCS) necessitates segregation from the B cell lymphoma, T cell lymphoma, Myeloid Sarcoma, Melanoma, Carcinoma, Dendritic and Histiocytic disorders such as a Plastic, Plasmacytoid or Dendritic cell neoplasm, Peripheral nerve sheath tumours or a Malignant Fibrous Histiocytoma and Langerhans Cell Histiocytosis [1]. Immune-histochemistry is advantageous in the analogy of various conditions.
| Bajaji A* Histopathologist in A B Diagnostics, India *Corresponding author: Anubha Bajaji, Histopathologist in A.B. Diagnostics, Histopathologist in A.B. Diagnostics, India, Tel: +995599957328; Email: | Review Article | |||
| Volume 3 Issue 1 | ||||
| Received Date: September 12, 2018 | ||||
| Published Date: September 21, 2018 |
Analysis and Attributes
Follicular dendritic cell sarcoma (FDCS) is a preponderantly adult disease with a median emergence at
44 years) [1]. Localized lesions (FDCS) progress mildly, with a mean survival of roughly 168 months (varying from 2 to 360 months). Limited reoccurrences and remote metastasis are proportionately at 27% and 28% respectively. Enhanced tumour expanse (> 6cm), the appearance of coagulative necrosis, a raised mitosis (> 5/10 hpf) and cytological aberrations may elucidate an inferior prognosis [7]. Tumour Stage may not influence the disease outcome. A two year survival for the preliminary or progressive, localized and distant metastasis is documented at 84.2%, 80% and 42.8% respective [1]. The neoplasm (FDCS) emerges as a gradual tumefaction, commonly situated in the head and neck or the abdominal lymph nodes. A limited cervical or an intra- abdominal swelling may appear in half the (50%) instances. The implicated extra-nodal sites are the Hepatic, Pulmonary, Tonsils and the Spleen [1]. The malignancy (FDCS) may be investigated with the contrast Computerized Tomography(CT) scans extending from the neck to the pelvis in order to assess adjunctive disease. Complete blood counts, bone marrow aspiration and biopsy are recommended. Concomitant viral evaluation such as Human Immune Deficiency Virus (HIV), Epstein Barr Virus (EBV) and Hepatitis assay may be accomplished [1]. Core needle biopsy or an Excision biopsy may be adopted for a precise identification of the tumefaction. Fine needle aspiration cytology (FNAC) may be circumvented.
Histopathology
The tumour specimen is composed of spindle to ovoid cells which may configure fascicles, whorls, diffuse sheets and cellular aggregates. Singular cells elucidate an indeterminate cellular outline and a moderate, non- discernible cytoplasm [Figures 1-9]. Nuclear pseudo- inclusions frequently appear along with bi-nucleation/ multi-nucleation of the tumour cells [3]. Electron microscopy depicts cytoplasmic prolongations and desmosomal intersections. Birbeck granules and plentiful liposomes may not be expressed [6]. Lympho-plasmacytic influx may emerge in majority (90%) of the instances [1] Reed Sternberg like cells may infrequently incite a misinterpretation of a Hodgkin’s Lymphoma. Immune- histochemistry is an advantageous technique to distinguish the tumour (FDCS) from the adjunct Histiocytic neoplasm. Follicular dendritic cell sarcoma is immune reactive to CD21, CD23, CD35, R4/23, Ki -FDC1p and Ki M4 with a variable quantification of CD68 [6]. Cluster in appears as a highly reactive molecule in the neoplasm (FDCS) and may be negative or weakly reactive in the complementary dendritic cell tumours [7, 8, 9, 10, 11, 12]. Desmoplakin. Vimentin, Fascin, Epidermal Growth Factor
Receptor (EGFR), CD45 and HLA-DR may be reactive [6]. Immunoglobulin and T cell receptor genes may be in a germ line disposition. Cytogenetic evaluation of the particular dendritic neoplasm (FDCS) is insufficient and may not benefit the interpretation [1].
Therapeutics
Surgical excision is the preferred modality. The majority (94%) of the patients with limited disease are treated with preliminary surgery [9]. Adjuvant therapy for excised localized lesions is controversial, though radiotherapy and chemotherapy may be employed. Concomitant Chemotherapy and Radiotherapy may benefit the protracted disease. Treatment regimens employed are cyclophosphamide/ vincristine/ doxorubicine, prednisone (CHOP), ifosamide, carbaplatin, etoposide(ICE) and doxorubicin/ bleomycin/ vincristine/ dacarbazine (ABVD), the therapies applicable to infiltrative lymphoma [4, 5, 6]. Cognitive lymphoma chemotherapy is recommended for diffuse follicular dendritic cell sarcoma (FDCS). The province of allogeneic bone marrow transplantation is indeterminate.
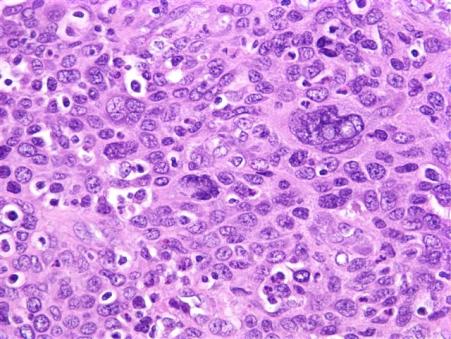
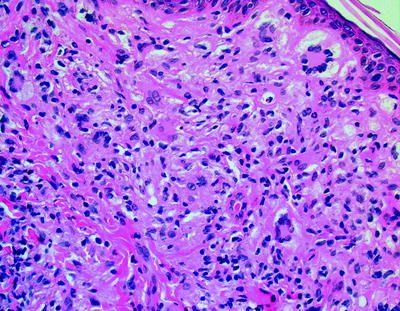
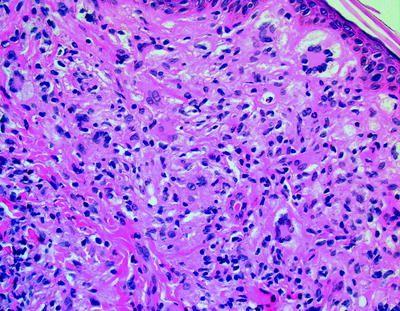
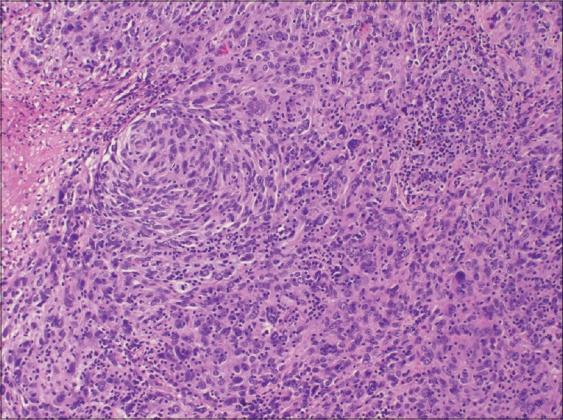
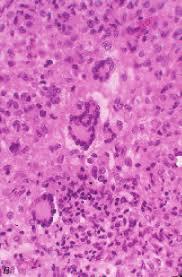
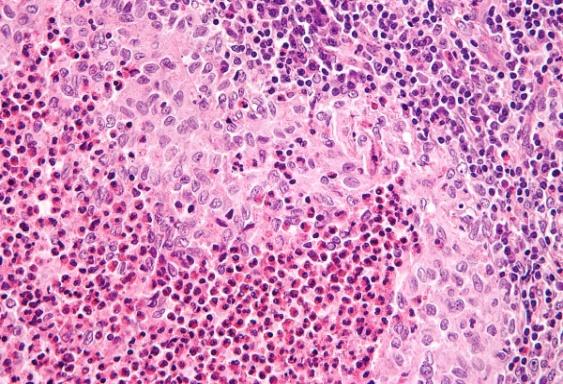
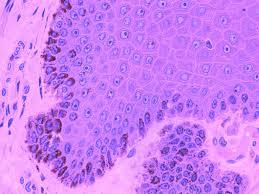
Langerhans Cell Histiocytosis (LCH): Morphology
The rampant proliferation of the langerhans cells, an immune accessory cell implicated in specific antigen capturing and delivery to the lymphoid cells is designated as Langerhans cell histiocytosis (LCH) [2]. The typical nuclei are irregular, generally elongated with multi- pronged, conspicuous grooves and convolutions. The cytoplasm is abundant and acidophilic (simulating an embryonal rhabdomyosarcoma). The langerhans cells are chiefly mononuclear though sporadic multinucleated configurations may be demonstrated. Langerhans cells and Langerhans cell histiocytosis (LCH)are immune reactive for S-100 protein, Vimentin, Langerin, (CD207), Fascin (a dendritic cell marker), CD1a, CD74 and HLA-DR besides the macrophage associated antigens CD68, Cathepsin D and Cathepsin [2]. They may not expound CD45RA, CD45B, CDw75, alpha 1 antitrypsin and epithelial membrane antigen (EMA). S100 protein, CD1a and Langerin evaluation is most beneficial. Electron Microscopy exemplifies a characteristic organelle, the Birbeck’s or Langerhans granule [2]. Langerhans cell histiocytosis (LCH) may appear as multitudinous disorder or a solitary lesion, confined to a single organ. The age of commencement is variable and extends from adolescents to the elderly. Lymph nodes may be implicated in the systemic kind of disease or it may a particular and preliminary manifestation of the disease.
Histopathology
The nodal sinuses are dilated with the mononuclear or multinuclear Langerhans cells admixed with a varying quantity of eosinophils. Focal necrosis with abutting eosinophils is frequent and may be restricted to the sinuses. The nodal configuration may be conserved or partially effaced. Specific organs exhibiting the disorder are the bone, lymph node, pulmonary, thymus, skin, central nervous system (CNS), gastric, hepatic, anus, female genital tract and the thyroid, especially in the Solitary form of Langerhans cell histiocytosis (LCH) [2]. Rosai Dorfman’s disease, parasitic infections, Kimura’s disease, Hypersensitivity reactions, Cat scratch disease , Erdheim –Chester disease, specific Malignant lymphoma, Hodgkin’s Lymphoma and Peripheral T cell lymphoma are the conditions which require a segregation from Langerhans cell histiocytosis [2]. Langerhans cell histiocytosis (LCH) is a disorder of obscure aetiology. A viral origin has not been proved. A disorder with the tumour cells depicting the ultra-structure and immune reactions of Langerhans cells has been described. It is designated as a Langerhans cell sarcoma, analogous to the inter digitating reticulum cell sarcoma and may be a malignant neoplasm of the accessory immune system cells [2].
Histiocytic Sarcoma: Morphology
Histiocytic Sarcoma (HS) is an infrequent, non langerhans cell disorder constituted of mature tissue histiocytes. Of obscure aetiology, the condition may concur with mediastinal germ cell tumour, implying that the Histiocytic Sarcoma (HS) may emerge from the pleuri- potent germ cells [1]. Follicular lymphoma, Myelo- dysplastic syndrome (MDS) and acute lymphoblastic lymphoma (ALL) may be concomitant with the Histiocytic Sarcoma (HS). A Trans –differentiation amidst the Histiocytic Sarcoma (HS) and Follicular Lymphoma cell clones along with the t (14:18) gene translocation and immunoglobulin heavy (Ig H) gene rearrangements, implicates an analogous commencement of the disorders. As chronic lymphocytic leukaemia (CLL)/small lymphocytic lymphoma (SLL) demonstrate clone specific immunoglobulin rearrangements, it is postulated that Histiocytic Sarcoma may develop [1]. Histopathology and Immune-histochemistry are essential in order to distinguish the condition. Immune reactivity for CD 163, CD 68 and lysozyme is exemplified.
Analysis and Attributes
An adult, male preponderance is noted (median presentation at 46-55 years), though no age is exempt. Solitary or multitudinous extra-nodal tumours are described, frequently in the gastro-intestinal tract, skin or soft tissues [1]. Disseminated lymph- node enlargement and manifold tumour loci may be exceptionally elucidated, with a poor prognosis. Fever, weight loss and compression of a vital structure (such as a small bowel obstruction) may descry systemic manifestations. Skin rash is exhibited at numerous sites with dermal lesions [3]. Haematological cytopenias may occur in one-third (30%) instances. A comprehensive staging with computerized tomography (CT) scans and a bone marrow aspiration and biopsy is mandated with solitary lesions which may exhibit a superior prognosis. Excision biopsy is the selected mode of investigation.
Histopathology
Enormous, propagating and dispersed cells twice the diameter of small lymphocytes may appear in concurrence with foci of spindle cell proliferation [1]. Vacuoles are demonstrated in the eosinophilic cytoplasm. Pleomorphic, eccentric nuclei may exhibit single or multiple nucleoli. A xanthomatous manifestation may ensue [3]. Histiocytic markers may be reactive such as CD163, CD 68 and lysozyme. CD 33 is usually non- reactive. S-100 protein may be weak or focally reactive. Ki 67 is variably depicted [1].
Therapeutics
Simple surgical excision or in combination with adjuvant radiotherapy or a surgical resection concomitant with adjuvant chemotherapy is the desirable mode of medication. Multi-various treatment options incorporated for a solitary, extra-nodal tumefaction remains to be evaluated. Cyclophosphamide/ vincristine/ doxorubicine, predinosone (CHOP) is a frequently employed regimen [1]. Adjuvant radiation therapy may diminish the on-site reoccurrences. Adjuvant chemotherapy may benefit the disseminated disease.
Interdigitating Dendritic Cell Sarcoma: Morphology
The typical inter-digitating dendritic cells (IDCs) are antigen presenting cells (APCs) situated in the T cell rich nodal paracortex. The cells proffer antigens to the T cells and monitor the cellular immune response [1]. The inter- digitating dendritic cells (IDCs) arise from the haematopoietic bone marrow precursors by modifying the langerhans cells during their migration through the lymph nodes (12). Malignant inter-digitating dendritic cells (IDCs) develop into the inter-digitating cell sarcoma (IDCS) which may react to S -100 proteins and Vimentin and may be non-reactive to CD1a and Langerin. Contrary to the Follicular dendritic cell sarcoma (FDCS) they may not exemplify CD21 and CD35. Inter-digitating cell sarcoma (IDCS) may accompany solid malignant disorders such as a B cell lymphoma, mycosis fungoides with dermal, hepatic, gastric, colon, mammary and brain tumefactions (1). Associated clones of inter-digitating cell sarcoma (IDCS) and low-grade B cell lymphoma may appear on account of trans-differentiation of the cellular lymphoid clones [4, 5]. Concordance of an inter-digitating cell sarcoma (IDCS) with a chronic lymphatic leukaemia/ small lymphocytic lymphoma (CLL/SLL) may ensue. Matching clones of Immunoglobulin H or Immunoglobulin K may accompany chronic 17 p deletions, as detected by Fluorescent in situ hybridization (FISH), thus indicating a similar ancestry of the malignant clones [1]. Equivalent disposition of the V J junction and trisomy 12 in the chronic lymphatic leukaemia (CLL) and inter-digitating cell sarcoma (IDCS) tumefaction imply a trans- differentiation of the lymphoma clones [5]. Contrary to the Follicular dendritic cell sarcoma (FDCS), a viral aetiology of inter-digitating cell sarcoma (IDCS) has not been elucidated. Inter-digitating cell sarcoma (IDCS) is predominantly non-reactive for Epstein barr virus (EBV) and the Human herpes virus8 (HHV8) genome [1]. Application of Calcineurin inhibitors (CNIs) may induce an inter-digitating cell sarcoma (IDCS), along with the T cells reaction to the antigen presenting inter-digitating cells (IDCs). Immune –histochemical and molecular techniques facilitate the determination of the disorder.
Analysis and Attributes
Inter-digitating cell sarcoma (IDCS) is an exceptional neoplasm. The tumour may usually be detected at 56.5 years (detection may extend from 21 months to 88years). The male to female proportion is 1.38:1. The clinical course fluctuates from a benign to an accelerated, fatal condition with dissemination. Instances of intra- abdominal participation and young patients with aggressive disease enunciate a poorer prognosis. Confined, limited lesions elucidate a 1 and 2 year continuance of 84.8% and 68.1% while the diffuse disorder depicts a 1 and 2 year survival of 38.5% and 15.8 % respectively [1]. A solitary lymph node tumefaction is frequent though the adjoining skin and soft tissue may be implicated [13]. Asymptomatic individuals may be elucidated although fatigue, fever and night sweats may ensue. The tumour staging incorporates a contrasting computerized tomography (CT) scan, extending from the neck to the pelvis for a comprehensive assessment. Complete blood counts, bone marrow aspiration and biopsy may be mandated. A viral aetiology has not been implied for the inter-digitating cell sarcoma (IDCS). Thus evaluation for Human immune deficiency virus (HIV) and an exhaustive Hepatitis spectrum is not a pre-requisite. Core needle biopsy or a selective Excision biopsy is required for the identification of the neoplasm. Fine needle aspiration cytology (FNAC) may be averted.
Histopathology
Classically, huge spindle to ovoid cells configuring whorls may be identified. The cells demonstrate a coarse nuclear chromatin with moderate to abundant cytoplasm and may simulate histiocytes. Small lymphocytes commingling with the enormous histiocytic cells is the essential attribute, although it is absent in carcinomas or sarcomas. Immune phenotype is non-reactive for CD1a with a reactive S100 protein, Vimentin, HLA DR, Fascin and CD45, with a variable CD68 and Lysozyme [1]. B cell marker CD20 and T cell markers CD3 and CD 5 may be non-reactive. Cytokeratin, Myeloperoxidase , CD1a , CD 21, CD 23, CD 30, CD 35, Clusterin, Langerin, CD34 , CD 79a, BCL 2 and BCL 6 may also be non reactive(3,13). Electron Microscopy depicts a lack of Birbeck’s granules. Immunoglobulin and T cell receptor genes appear in a germ line configuration.
Therapeutics
The preliminary treatment for limited disease may be radiotherapy or surgical excision, as it enhances the longevity. Diffuse disease may be managed by chemotherapeutic options such as cyclophosphamide/ vincristine/ doxorubicine, predinosone (CHOP), ifosamide, carbaplatin, etoposide(ICE) and doxorubicin/ bleomycin/ vincristine/ dacarbazine (ABVD) with modifiable outcome [13]. The therapeutic options for the disseminated lesions, however, lack concurrence.
Indeterminate Dendritic Cell Sarcoma (INDCS)
Morphology: of the indeterminate dendritic cell sarcoma or the indeterminate cell histiocytosis is an exceptional, malignant proliferation of the dendritic accessory cells situated in the dermis. The indeterminate cells may be analogous to the morphology and immune-phenotype of the Langerhans cells (apart from the ultra-structural Birbeck’s granules) .Thus the indeterminate cells may indicate the mature configuration of Langerhans cells. The indeterminate cell propagation may be concordant with nodular scabies, pityriasis rosea and low grade B cell lymphoma [1]. Immune phenotypic markers may be reactive for S-100 protein and CD 79 1a. Birbeck’s granules are absent on electron microscopy [1].
Analysis and Attributes
The intermediate dendritic cell sarcoma (INDCS) usually elucidates solitary or multiple papules, nodules or plaques appearing on the trunk, face, neck and extremities in an indeterminate pattern. A skin biopsy is generally diagnostic. Computerized tomography (CT) scans and bone marrow biopsy may be mandated.
Histopathology
Microscopy exhibits an indiscriminate dermal infiltrate. The cellular component is comprised of cells with irregular nuclear grooves and clefts similar to the langerhans cells [1] the cytoplasm is abundant, pale and eosinophilic. Multinucleated giant cells may be demonstrated along with partial spindling or a dendritic configuration. Birbeck granules and desmosomes are absent on electron microscopy, though inter-digitating cell processes may be detected. Immune phenotypic markers describe the tumour cells to be reactive for s100 protein and CD 1a. Non -reactivity for b cell and t cell markers, CD 30, CD 163 CD21, cd23, CD 35 and langerin. Factor xiii a and CD34 may be deficient, in contrast to xantho-granuloma and dermato-fibrosarcoma protuberans respectively [1]. CD45, CD 68, lysozome and CD4 are variably reactive. The human androgen receptor gene assay may be able to clone the intermediate dendritic cell sarcoma (INDCS).
Therapeutics
The tumefaction is gradual and indolent. Recent, fresh lesions may ensue with autonomous retrogression of the tumour. Surgical resection is the preferred treatment modality. Outcomes with chemotherapy and radiotherapy may be unreliable, particularly with diffuse disease. Multiple therapeutic options may be contemplated [1].
Fibroblastic Reticular Cell Tumour: Morphology
Fibroblastic reticular cell tumour (FRCT) is an exceptional neoplasm of the fibroblastic reticular cells which configure the abutting cells of the stroma, are established in the para-follicular zones and the inmost cortex of the lymph node and accompany the nodal reticular groundwork. Intercommunication of the inter- digitating dendritic cells (IDCs) and T cells in the primary immune response may be facilitated by the fibroblastic reticular cells. Fibroblastic reticular cell tumour (FRCT) and the cytokeratin positive interstitial reticular cell tumour may be identical. Fibroblastic reticular cell tumour (FRCT) appears in the lymph node, though it may emerge in sites such as the spleen, pulmonary, hepatic or soft tissue [1]. The concordance of smoking, drug abuse and viral infection with fibroblastic reticular cell tumour (FRCT) is debatable. Fibroblastic reticular cell tumour (FRCT) may be separated from the inter-digitating cell sarcoma (IDCS) and follicular dendritic cell sarcoma (FDCS) based on the immune marker reactions (IHC). Immune reactivity with Vimentin, Smooth muscle actin, Factor XIIIa and Desmin is described. The tumour is non- reactive for CD21, CD35 and CD1a. Immune reaction profile aids the demarcation of the tumour from inter digitating cell sarcoma (IDCS) and follicular dendritic cell sarcoma (FDCS) [1].
Analysis and Attributes
A male predilection and a median age of emergence at 61 years is exhibited. Cervical and mediastinal lymph nodes are frequently implicated. Extra nodal tumours are situated at the hepatic, spleen, renal, adrenal, bone and soft tissue sub-sites [1]. Patients with higher stage disease Aggressive tumours demonstrate a diminished survival. Limited disease elucidates a 2 year continuance at 85.7 % while with the disseminated disease the 2 year median life expectancy was 15 months [1]. A recent, asymptomatic tumefaction is perceived which may be surgically removed. Multitudinous, expanded lymph nodes may be evaluated by a Computerized Tomography (CT) scans and a bone marrow biopsy. The value of these procedures in a solitary nodal incrimination is indeterminate. Excision biopsy is the selective mode of detection [1].
Histopathology
Spindle to ovoid cells articulating whorls in the paracortex, accompanied by abundant, reticulated fibrous tissue may be enunciated. Vimentin, Desmin, Factor XIIIa, Smooth muscle actin are immune –reactive. CD 45RB, CD21, CD35, S-100 protein, CD65 and CD 1a are usually not delineated [1, 3]. Marginal, fusiform , elongated dendritic cytoplasmic projections and desmosomal intercellular junctions may be exhibited on ultra- structure.
Therapeutics
Surgical eradication is preferred for localized lesions. Adjuvant radiotherapy may be of limited value. Adjuvant chemotherapy has a minimal benefit in the controlled disease. Treatment options for disseminated fibroblastic reticular cell tumour (FRCT) have been insufficiently analysed though clinical trials may be a pre-requisite.
Disseminiated Juvenile Xantogranuloma (DJX)
A histocytic proliferation identical to the dermal juvenile xanthogranuloma (JXG) is the hallmark of the disorder. Singular dermal xanthogranulomatous lesions (JXG) are frequent with negligible advancement to the distant types. Skin lesions generally recede, though tumours may appear in the brain, soft tissue or with the diffuse disorder [1, 2]. Type I neurofibromatosis or juvenile myelomonocytic leukemia may co-exist with juvenile xanthogranuloma [3]. Concomitant langerhans cell histiocytosis (LCH) and juvenile xanthogranuloma (JXG) may arise, thereby indicating an identical cellular clone of emergence.
Analysis and Attributes
Disseminated juvenile xanthogranuloma (DJX) presents at 10 years of age with one half of the lesions appearing in infancy. Skin, soft tissue with upper airway mucosa is frequent sites of representation. Solitary papules or miniature, multiple lesions may ensue [14]. The central nervous system (CNS), pulmonary, hepatic or ocular sites with lymph nodes and bone marrow may be implicated [1]. Diabetes insipidus, seizures, hydrocephalus and alterations in the mental state may be demonstrated with central nervous system lesions. Excision biopsy of the tumour with appropriate immune histochemical evaluation is recommended. Staging computerized tomography (CT) scans and bone marrow biopsy persist as indeterminate options.
Histopathology
The xanthogranulomatous (JXG) component is comprised of a miniature, round to ovoid cell with a bland, uniform nucleus and stained cytoplasm. Touton giant cells are elucidated at the dermal sites, though infrequent in the non dermal locations. Xanthomatous and inflammatory elements may be exhibited. [1, 3]. Vimentin, Lysozyme, CD14, CD68, CD 163, Stabilin -1 and co-factor XIIIa may be immune-reactive. CD1a and S 100 protein are usually non-reactive though S-100 may be weakly or focally positive [1]. Immunoglobulin G and T cell receptor genes may configure in a germline manner. A multifocal appearance may resemble a lymphoma [15].
Therapeutics
Cutaneous, subcutaneous and soft tissue juvenile xanthogranuloma (JXG) may not require intervention as the lesion automatically regresses. A central nervous system (CNS) or a symptomatic lesion may require additional evaluation and adjuvant chemotherapy. Treatment regimens with vincristine, prednisone and methotrexate may be employed though the patients or the nodules of langerhans cell histiocytosis may demonstrate an inconstant outcome. Additional evaluation in the form of clinical trials may be beneficial [Table 1, 2].
| FDCS & IDCS | FRCT | HS | INDCS | JXG | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Anaplastic Large cell lymphoma ( FDCS for IDCS, IDCS for FDCS) | FDCS | Anaplastic Large cell lymphoma Diffuse Large B cell lymphoma | LCH | Dermatofibroma | ||||||||||
| Inflammatory pseudotumour | IDCS | Haemophagocytic Lympho-histiocytosis | Pitryasis rosea | Eruptive Xanthoma | ||||||||||
| Intranodal Myofibroblastoma | INDCS | LCH | Scabies | LCH | ||||||||||
| Non Hodgkin’s Lymphoma | Palisaded Myofibroblastoma | Lymphoma | Tcell lymphoma9cutaneous T cell hyperplasia, mycoises fungoides) | Mastocytoma Papular xanthoma | ||||||||||
| Peripheral nerve sheath tumour | Sarcoma | Metastatic Carcinoma | Spitz nevus Tuberous Xanthoma | |||||||||||
| True histiocytic lymphoma | Melanoma | Xanthoma dissemination | ||||||||||||
| Clinical Features | FDCS | IDCS | INDCS | HS | FRCT | |||||||||
| Slowly evolving mass, generally a lymph node | Asymptomatic Solitary lymph node mass | DermalPapules Nodules Plaques | Solitary mass with systemic symptoms Rash , skin lesions | Asymptomatic mass | ||||||||||
| Histology | Spindle to oval cells with whorls | Spindle to oval cells with whorls | Resemble langerhans cells with irregular nuclear grooves and clefts | Large round to oval cells with foci of spindling | Spindle to ovoid cells with whorls in the paracortex | |||||||||
| Immune Markers | CD4+ CD21+CD34- CD35+CD68+/- Fascin + | CD4+CD45+/- CD68+ Fascin+ S100+ | CD1a-CD4+ Fascin + S100+CD68+/- Birbeck’s granules- | CD163+CD68+, Lysozyme + CD1a – CD21- CD35- CD 33- | Vimentin + Desmin +Smooth muscle actin + Factor XIIIa+ CD21- CD35-S100- CD1a- | |||||||||
| Treatment for limited disease | Surgical Resection with adjuvant Chemotherapy and Radiotherapy | Surgical Resection with adjuvant Radiotherapy | Surgical Resection | Surgical Resection with adjuvant Radiotherapy | Surgical Resection with adjuvant Radiotherapy | |||||||||
| Treatment for disseminated disease | Lymphoma like chemotherapy | Lymphoma like chemotherapy | Mutimodality treatment regimen | Lymphoma like chemotherapy | Participation in a clinical trial |
Table 2: Accessory diagnosis - dendritic cell sarcoma.
References
-
Samir Delia, Shao H, Sagatys E, Cualing H, Sokol L (2004) Dendritic cell and histiocytic neoplasm: Biology, Diagnosis and Treatment Cancer control 21(4): 290-300.
-
Rosai and Ackerman’s Surgical Pathology tenth (Edn) PP-1803.
-
World health organization (2008) World health organization of classification of Haematopoietic and Lymphoid tissues, Lyon France; International agency for research on cancer: 2008/2016.
-
Feldman AL Arber DA, Pittaluga S, Martinez A, Burke JS, et al. (2008) Clonally related follicular lymphomas and histiocyte/dendritic cell sarcomas: evidence of trans-differentiation of the follicular lymphoma clone BLOOD 111(12): 5433-5439.
-
Fraser CR, Wang W, Gomez M, Zhang T, Mathew S, et al. (2009) Transformation of Chronic Lymphocytic Leukaemia /Small Lymphocytic Lymphoma to interdigitating dendritic cell sarcoma: evidence of trans-differentiation of the lymphoma clone Am J Clin Pathol 132(6): 928-939.
-
Steven H Swerdlow, Campo E, Pileri SA, Harris NL, Stein H, et al. (2016) The 2016 revision of world health organization classification of lymphoid neoplasm BLOOD 127(20): 2375-90.
-
Grogg KL, Macon WR, Kurtin PJ, Nascimento AG (2005) A survey of clustering and fascin expression in sarcoma and spindle cell neoplasm: strong Clusterin immune-staining is highly specific for follicular dendritic cell tumour Mod Pathol 18(2): 260-266.
-
Shia J, Chen W, Tang LH, Carlson DL, Qin J, et al. (2006) Extranodal Follicular dendritic cell sarcoma: Clinical Pathologic and Histogenetic characteristics of an under-recognized disease entity” Virchow’ Arch 449(2):148-158.
-
Perkins SM, Shinohara ET (2013) Interdigitating and Follicular Dendritic cell sarcoma: a SEER analysis” Am J Clin Pathol 36(4): 395-398.
-
Soriano AO, Thompson MA, Admirand JH, Fayad LE, Rodriguez AM, et al (2008) Follicular dendritic cell sarcoma: a report of 14 cases and a review of literature Am J Haematol 82(8): 725-728.
-
Li L, Shi YH, Guo ZJ, Qiu T, Guo L, et al (2010) Clinico- pathologic features and prognosis assessment of extranodal follicular dendritic cell sarcoma World J of Gastroenterol 16(20): 2504-2519.
-
Wu L, Lui YJ (2007) Development of dendritic cell lineages Immunity 26(6): 741-750.
-
Jiang YZ, Dong NZ, Wu DP, Xue SL (2013) Interdigitating dendritic cell sarcoma presenting simultaneously with acute myelomonocytic leukaemia: report of a rare case and literature review Int J Haematol 97(5): 675-666.
-
Zeng W, Meck J, Cheson BD, Ozdemirli M (2011) Histiocytic sarcoma trans-differentiation from follicular lymphoma presenting as a cutaneous tumour J Cutan Pathol 38(12); 999-1003.
-
Maly E, Przyborska M, Rybczyńska A, Konatkowska B, Nowak J, et al. (2012) Juvenile Xanthogranuloma with clonal proliferation of the bone marrow: J Paediatr Haematol Oncol 2012: 34(3); 222-225.
-
Figure1 Courtesy: Hemepath
-
Figure 2 Courtesy: Clinical cancer investigation journal.
-
Figure 3 Courtesy: Pathology Outlines.
-
Figire 4 Courtesy: Diagnostic pathol biomed central.
-
Figure 5 Courtesy: Springer-link.
-
Figure 6 Courtesy: Wikipedia.
-
Figure 7 Courtesy: Med cell yale edu.
-
Figure 8 Courtesy: Web-pathology.
-
Figure 9 Courtesy: i-Path Network
- Management of Gallbladder Perforations: A Review
- From The Mouth to the Gut: The Oral Microbiome's Role in Promoting Gastrointestinal Disease
- Case Report: Intraductal Papillary Mucinous Neoplasm (IPMN) Complicated by Portal Vein Plaquing and Biliary Obstruction Mimicking Pancreatic Metastatic Malignancy
- Management of Non-Cirrhotic Portal Hypertension during Pregnancy: A Review
- Effectiveness of Omeprazole versus Pantoprazole for Symptomatic Relief of Gastro-Esophageal Reflux Disease (GERD)/ Acid Peptic Disease (APD): A Real-World Evidence (RWE) Study
- Case of Splenic Infarction; A Rare Presentation of Complicated Enteric Fever in a Pediatric Patient