In-Silico Identification of Potent Inhibitors of COVID-19 Main Protease (Mpro) from Natural Products
COVID-19 is rapidly spreading and there are currently no specific clinical treatments available. The absence of an immediate available vaccine against SARS-CoV-2 made it hard for health professionals to tackle the problem. Thus, the need of ready to use prescription drugs or herbal remedies is urgent. SARS-CoV-2 main protease (Mpro) protein structure is made available to facilitate finding solutions to the present problem. In this brief research, we compare the efficacy of some natural compounds against COVID-19 Mpro to that of Hydroxy-Chloroquine in silico. Molecular docking investigations were carried out using AutoDock. Virtual screening was performed using AutoDock Vina and the best ligand / protein mode was identified based on the binding energy. Amino Acids residues of ligands interactions were identified using free version of Discovery Studio Visualizer and PyMOL. According to present research results, Gallic acid, Quercetin, Hispidulin, Cirsimaritin, Sulfasalazine, Artemisin and Curcumin exhibited better potential inhibition than Hydroxy-Chloroquine against COVID-19 main protease active site. Our provided docking data of these compounds may help pave a way for further advanced research to the synthesis of novel drug candidate for COVID-19.
Introduction
Coronaviruses are a large family of enveloped, RNA viruses. There are 4 groups of coronaviruses: alpha and beta, originated from bats and rodents; and gamma and delta, originated from avian species [1]. Coronaviruses are responsible for a wide range of diseases in many animals, including livestock and pets [2]. In humans, they were thought to cause mild, self-limiting respiratory infections until 2002, when a beta-coronavirus crossed species barriers from bats to a mammalian host, before jumping to humans, causing the Severe Acute Respiratory Syndrome, SARS, epidemic. More recently, another beta-coronavirus is responsible for the serious Middle East Respiratory Syndrome, MERS, started in 2012 [3]. The novel coronavirus responsible for the Coronavirus Disease 2019 pandemic, COVID-19, is also a beta-coronavirus [4]. The genome of the virus is fully sequenced and appears to be most similar to a strain in bats, suggesting that it also originated from bats. The virus is also very similar to the SARS-coronavirus and is therefore named SARS-coronavirus 2, SARS-CoV 2 [5]. In order to infect a host cell, the spikes of the virus must bind to a molecule on the cell surface. The novel coronavirus appears to use the same receptor as SARS-coronavirus for entry to human cells, and that receptor is the angiotensin-converting enzyme 2, ACE2 [4]. Infection usually starts with cells of the respiratory mucosa, and then spreads to epithelial cells of alveoli in the lungs. Receptor binding is followed by fusion of the viral membrane with host cell membrane, and the release of nucleocapsid into the cell. Currently, no specific clinical therapies are available for the treatment of SARS-CoV-2 mediated infections [6]. Thus, the need of the hour is the identification and characterization of a new drug candidate to inhibit binding the COVID-19 main protease (Mpro). The Mpro plays an essential role in the virus replication process. It cleaves the pp1a and pp1b polyproteins, to release functional proteins including RNA polymerase, endoribonuclease and exoribonuclease. Therefore, inhibition of Mpro activity could stop the spread of infection, as the released crystal structure of Mpro (6lu7) was obtained by crystallization with a peptide like type inhibitor (N3). The enzyme has a molecular weight of 33.79 kDa and forms a dimer, where each monomer has three domains: domain I (residues 8–101) domain II (102– 184) consists of an antiparallel beta, and domain alpha III (residues 201–301). Thus, the His 41 and Cys145 catalytic ports located between domains I and II, while the amino acids, Thr24, Leu27, His41, Phe140, Cys145, His163, Met165, Pro168 and His172 form a hydrophobic environment in the pocket [7].
To this aims, we have screened in silico the interaction between the main protease COVID-19 (Mpro) active site with natural compounds that displays a large variety of biological activities.
Experimental Design, Materials, and Methods
Computational chemistry or as known as molecular modeling is a fascinating branch of chemistry. It uses modeling and virtual simulations to help solve chemistry modern problems. Lately, virtual screening of compound libraries has become a standard technology in modern drug discovery pipelines [8]. In our study, to perform in-silico specific site docking, we used a powerful bioinformatics tool; AutoDock Tools-1.5.6. In order to visualize the data, we utilized a free version of MOE software (Molecular Operating Environment) and PyMOL software.
Protein Selection and Preparation
The complete genome of the main protease of COVID-19 was retrieved from PDB. PDB ID: 6LU7. The downloaded structures were prepared prior to docking as fellow: First, we visualized the PDB file in PyMOL then removed Hetatms and kept only Chain A. Next, we optimized hydrogen bonds structures and added atoms in missing loops or side chains. Finally, we removed water molecules and saved our files in a PDB file format.
Ligand Preparation
The structures of our ligands were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov) and saved in SDF format. Files were converted from SDF to PDB format using PyMOL.
Molecular Docking
For 6LU7 the center of active site of the grid was determined according to the position of peptide like inhibitor N3 in the structure [7]. The coordinates of the position are X: -16.308 Y: 11.57, and Z: 72.881 at grid spacing of 0.500 Angstrom. Virtual screening was carried our using AutoDock Vina [9] and the best ligand / protein mode was identified based on the binding energy. The scoring function of AutoDock Vina is: C=∑i<jftitj(rij), where the summation is over all of the pairs of atoms that can move relative to each other, normally excluding 1–4 interactions, i.e. atoms separated by 3 consecutive covalent bonds. Here, each atom i is assigned a type ti, and a symmetric set of interaction functions ftitj of the interatomic distance rij should be defined [9].
Pharmacophore Mapping
Pharmacophore, represents the spatial arrangement of features that is essential for a molecule to interact with a specific target receptor, is an alternative method despite molecular docking for achieving this goal. In this study, the pharmacophore mapping is carried out for the Gallic Acid the best ligand among the selected ligands using Free Version of Discovery Studio Visualizer.
In silico ADME and Predicted Bioactivity Study
Physiochemical and toxicological studies were conducted under SwissADME online software and Molinspiration online software (Table 3). The SMILES structures of ligands were obtained from PubChem database. The software allows us to compute and predict ADME parameters (Absorption Distribution Metabolism Excretion). Pharmacokinetic properties, “druglike”, nature and medicinal chemistry friendliness of molecules. Simulation of physiochemical and toxicological behavior of our ligands was obtained from SwissADME developed by the Molecular Modeling Group from the Swiss Institute of bioinformatics (http:// www.swissadme.ch/index.php). The parameters obtained are: Molecular Weight (g/mole), H-bond donors, H-bond acceptors, Lipophilicity (Log Po/w), Water Solubility (Log S), Molar Refractivity, Gastro Intestinal GI absorption, and Blood-Brain-Barrier BBB permeability. Predicted bioactivity parameters were completed from Molinspiration online software developed by Bratislava University (https:// www.molinspiration.com). The parameters obtained are:
G Protein-Coupled Receptor GPCR ligand; Ion channel modulation; Kinase inhibitor; Nuclear receptor ligand; Protease inhibitor and Enzyme inhibitor.
Data
In this research work: Figure 1. Shows the PDB ID, resolution and description of COVID-19 main protease
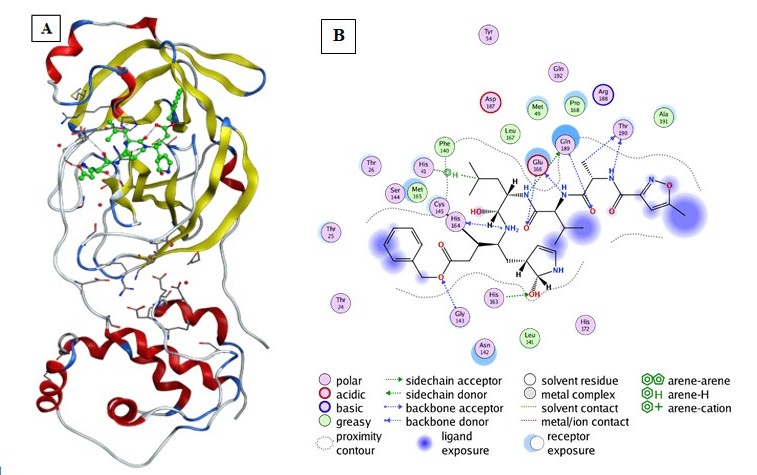
selected for this study. Table 1 provides the structure of chosen ligands. Table 2 gives docking results of COVID-19 main protease 6LU7. Table 3 represents the results the predicted physiochemical and biological activity of assessed molecules. The 3D interactions of the high scored ligands with COVID-19 main protease active sites are shown in Figures 2-10.
Figure 1: Structure representation of COVID-19 main protease (Mpro) in complex with an inhibitor N3. (A)-Representation of the crystal structure of COVID-19 main protease in complex with an inhibitor N3. (Yellow color: Mpro domain I, Bleu color: Mpro domain II, Red color: Mpro domain III. Green color: The peptide like inhibitor N3, Gray color represents coils). (B) 2D interaction of the peptide like inhibitor N3 with amino acid residues of Mpro COVID-19. (Generated using Free Version of Discovery Studio Visualizer).
| Name of ligand | Structure of ligand | Name of ligand | Structure of ligand |
|---|---|---|---|
| Quercetin | Chemical structure of Quercetin | Hispidulin | Chemical structure of Hispidulin |
| Cirsimaritin | Chemical structure of Cirsimaritin | Artemisin | Chemical structure of Artemisin |
| Curcumin | Chemical structure of Curcumin | Hydroxy-Chloroquine | Chemical structure of Hydroxy-Chloroquine |
| Thymoquinone | Chemical structure of Thymoquinone | Gallic Acid | Chemical structure of Gallic Acid |
| Sulfasalazine | Chemical structure of Sulfasalazine |
Table 1: Name of ligand and structure (https://pubchem.ncbi.nlm.nih.gov).
Results
Results of Binding Affinities of the Ligands into COVID-19 Main Protease (6LU7) Active Site
The binding energies obtained from the docking (AutoDock Vina) of the active site of COVID-19 main protease 6LU7 were presented in Table 2.
Gallic Acid, Quercetin, Hispidulin, Cirsimaritin, Sulfasalazine, Artemisin and Curcumin showed best binding energy to 6LU7 active site than that of Hydroxy-Chloroquine (Table 2). Gallic Acid: exhibited the first-lowest binding energy to 6LU7 (Binding energy to 6LU7= -8.3 kcal/mol). As shown in Table 2, Figure 2. Gallic Acid was well fitted into the active pocket of 6LU7. Gallic Acid formed hydrogen bonds with Ser144, Cys145, His163, Glu166, and Gln189. Furthermore, the aromatics groups of Gallic Acid were found to be interacting with Met165 and Cys145 via aromatic interaction. Gallic Acid aliphatic groups will be responsible for the formation of Van der Waals interactions.
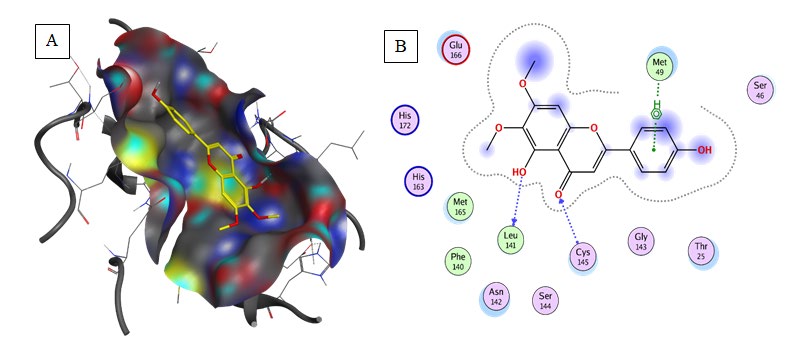
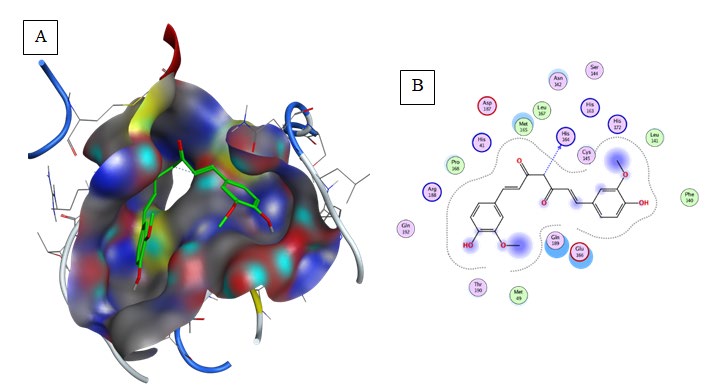
Quercetin: exhibited the second lowest binding energy to 6LU7(Binding energy to 6LU7= -7.5 kcal/mol). As shown in Table 2, Figure 3. Quercetin was well fitted into the active pocket of 6LU7. Quercetin formed hydrogen bonds with Leu141 and His163. Hydrogen bond interaction might be due to the 7 H-bond acceptors of Quercetin. As shown in figure 3 the aromatics groups of this flavonoid were found to be interacting with Glu166, Cys145, Met165, and Met49 via a variant of aromatic interaction. Furthermore, Quercetin aliphatic groups will be responsible for the formation of Van der Waals interactions.
Hispidulin (Binding energy to 6LU7= -7.3kcal/mol): exhibited the third-lowest binding energy at the active site of COVID-19 main protease Table 2. Hispidulin was well fitted into the active pocket of 6LU7 and it formed hydrogen bonds with His163, Leu141, Ser144, and Cys145 Figure 4. Furthermore, the aromatics groups of Hispidulin were found to be interacting with Met49 and Cys145 via aromatic interaction. Also, Hispidulin interacted with Glu166 and Phe104 via carbon-hydrogen bonds. Moreover, Hispidulin
aliphatic groups will be responsible for the formation of Van der Waals interactions, which compose a relatively hydrophobic environment.
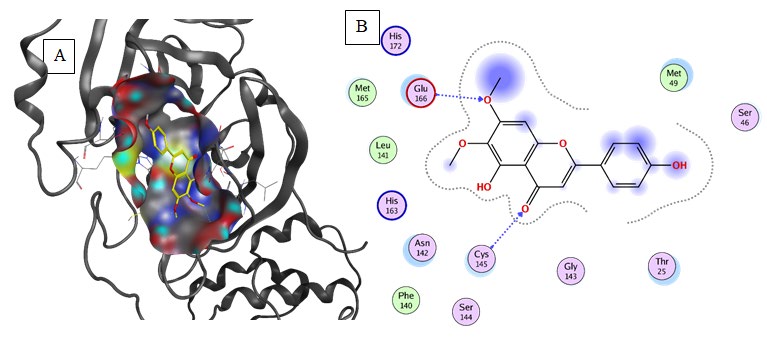
Cirsimaritin (Binding energy to 6LU7= -7.2kcal/mol): Predicted results illustrate that 6LU7 critical binding residue; Glu166, Cys145, and Ser144 form hydrogen bonds with Cirsimaritin. The His163 and Glu166 residues interact with Cirsimaritin via carbon-hydrogen bonds. The aromatics groups of Cirsimaritin were found to be interacting with Met49 and Cys145 via aromatic interaction. Furthermore, the hydrophobic environment was composed of the aliphatic groups which are responsible for the formation of Van der Waals interactions (Table 2, Figure 5).
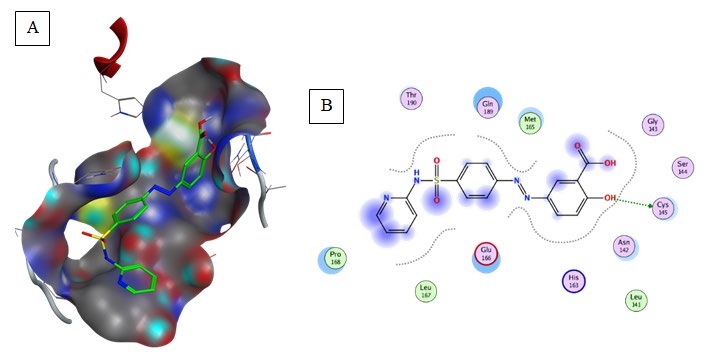
Sulfasalazine (Binding energy to 6LU7= -7.2 kcal/ mol): Amino acids predicted for Sulfasalazine binding in COVID-19 main protease were: Gly143, Ser144, Cys145, Met165, Leu167, and Pro168 as shown in Table 2, Figure 6. Sulfasalazine form hydrogen bonds with Gly143, and Ser144, and aromatic interaction with Cys145, Met165, Leu167, and Pro168. An amount of van der Waals interactions were composing a relatively hydrophobic environment.
Artemisin (Binding energy to 6LU7= -6.8 kcal/mol): Hydrogen bonding was predicted between 6LU7 actives sites His163 & Glu166 and the hydroxy functional group as shown in Table 2, Figure 7. Furthermore, van der Waals interactions were composing a relatively hydrophobic environment.
Curcuma (Binding energy to 6LU7= -6.8 kcal/mol): Hydrogen bonding was predicted between Glu 166 and the hydroxy group of the ligand (Table 2, Figure 8). A Pi-Sulfer and Pi-Alkyl interactions were predicted between Cys145 and Met 165 with the aromatic group of the compound. Carbon-hydrogen bond and van der Waals interactions were predicted between Gln189 and Glu166 with the aliphatic groups of Curcuma.
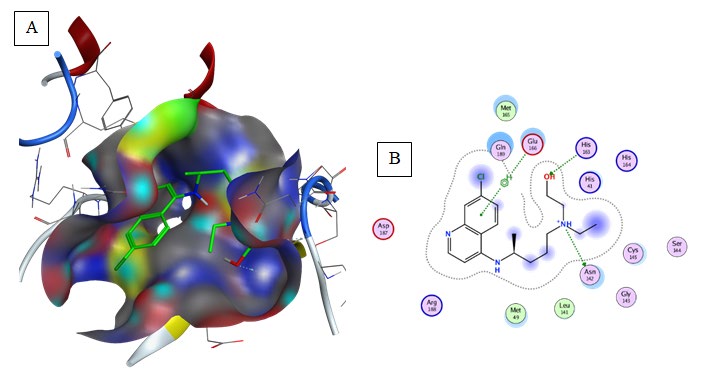
Hydroxy-Chloroquine (Binding energy to 6LU7= -5.9 kcal/mol): Hydrogen bonding was predicted between His164 and the compound (Table 2, Figure 9). Hydroxy- Chloroquine aromatic groups were responsible for the formation of aromatic interaction with Met165 and His41. Carbon-hydrogen bond and Alkyl interaction were formed between Leu141, Cys145, and His14 within the aliphatic groups composing a relatively hydrophobic environment.
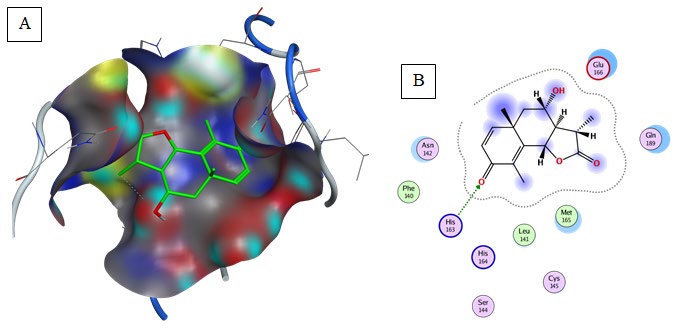
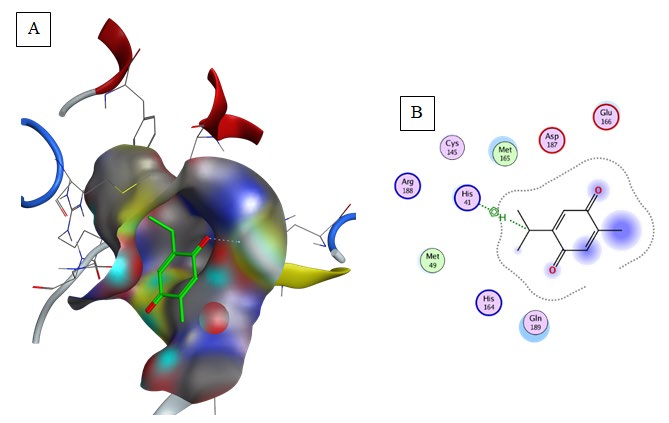
Thymoquinone (Binding energy to 6LU7= -5.1 kcal/mol): Pi-Alkyl interaction was predicted between Met165 and the aromatic group of the compound (Table 2, Figure 10).
| Ligand | Vina score (kcal/mol) | Receptor | Interaction | Distance (Å) | E (kcal/mol) |
|---|---|---|---|---|---|
| Gallic Acid | -8.3 | N CYS 145 | H-acceptor | 3 | -1.8 |
| N GLU 166 | pi-H | 4.54 | -1.3 | ||
| OE1 GLN 189 | H-donor | 2.7 | -3.4 | ||
| Quercetin | -7.5 | OE2 GLU 166 | H-donor | 2.6 | -3.6 |
| O PHE 140 | H-donor | 2.72 | -1.5 | ||
| ND2 ASN 142 | H-acceptor | 3.41 | -0.8 | ||
| NE2 HIS 163 | H-acceptor | 2.87 | -1.7 | ||
| O LEU 141 | H-donor | 3.17 | -1.6 | ||
| Hispidulin | -7.3 | N CYS 145 | H-acceptor | 3.13 | -2 |
| CE MET 49 | pi-H | 3.64 | -0.6 | ||
| Cirsimaritin | -7.2 | N CYS 145 | H-acceptor | 3.12 | -1.9 |
| N GLU 166 | H-acceptor | 3 | -0.8 | ||
| Sulfasalazine | -7.2 | SG CYS 145 | H-donor | 3.59 | -1.9 |
| Artemisin | -6.8 | NE2 HIS 163 | H-acceptor | 3.2 | -2.2 |
| Curcuma | -6.8 | O HIS 164 | H-donor | 3.22 | -0.9 |
| Hydroxy- Chloroquine | -5.9 | OD1 ASN 142 | H-donor | 3.36 | -1.9 |
| NE2 HIS 163 | H-acceptor | 3.18 | -1.4 | ||
| N GLU 166 | pi-H | 4.73 | -0.9 | ||
| Thymoquinone | -5.1 | 5-ring HIS 41 | H-pi | 3.67 | -0.8 |
Table 2: The hydrogen bond energy of the Gallic Acid, Quercetin, Hispidulin, Cirsimaritin, Sulfasalazine, Artemisin, Curcuma, Hyd
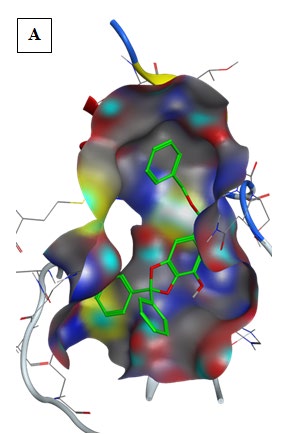
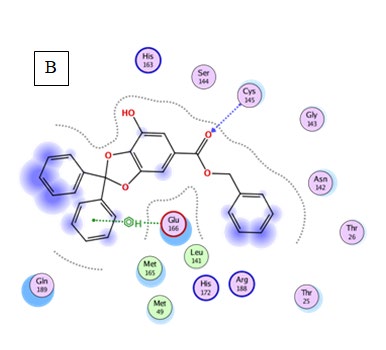
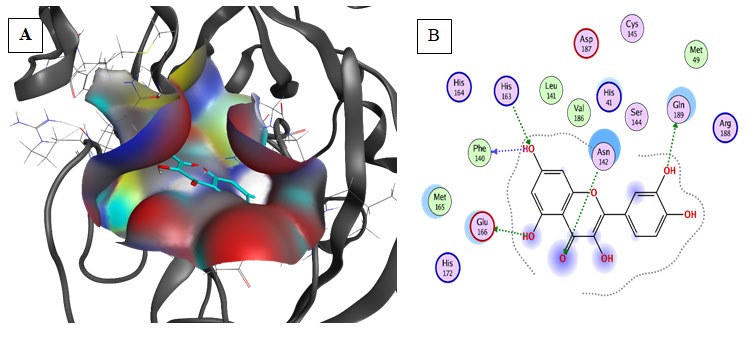
Results of Pharmacophore Study
The pharmacophore mapping is carried out for the Gallic Acid the best ligand among the selected ligands.
The Gallic Acid showed nine chemical features including 3 Hydrogen bonds acceptor, 1 Hydrogen bonds donor, 1 Hydrophobic groups and 4 Aromatic rings (Figure 11A).
The chemical features of Gallic acid attribute a strong biological activity to the molecule as a protease inhibitor,
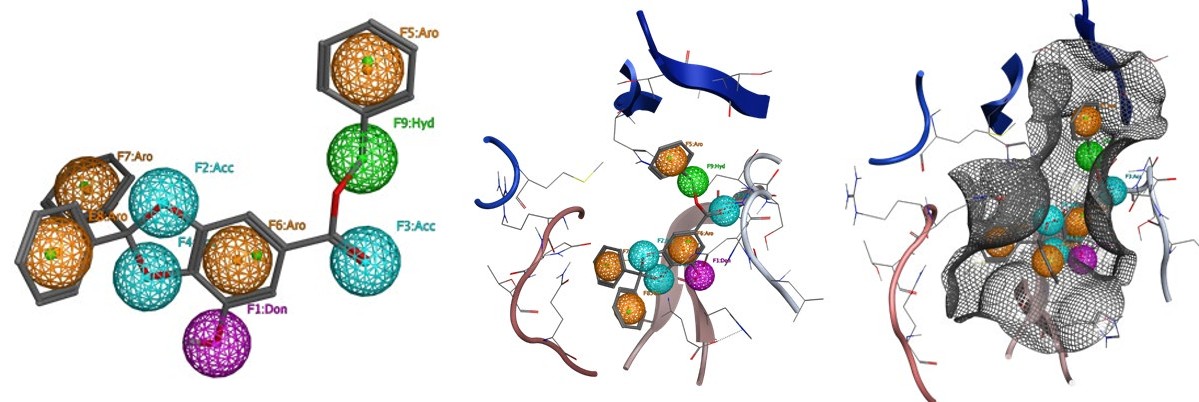
enzyme inhibitor, kinase inhibitor and nuclear receptor ligand (Table 3).
According to in silico results, Hydrogen bonds acceptor, and Hydrogen bonds donor formed hydrogen bonds with Ser144, Cys145, His163, Glu166, and Gln189. Furthermore, the aromatics rings of Gallic Acid were found to be interacting with Met165 and Cys145 via aromatic interaction. Gallic Acid Hydrophobic groups will be responsible for the formation of Van der Waals interactions.
Results of in Silico ADME and Predicted Bioactivity Study
assessed molecules conforms to Lipinski’s rule of five [10], (hydrogen bond donors ≤5; hydrogen bond acceptors ≤10; Molecular weight ≤ 500 Daltons; Octanol-water partition coefficient (log P) ≤5). According to the obtained results all the assessed drugs are safe for human use.
According to the in silico ADME study (Table 3) all
| Names of molecules | PubChem CID | Molecular Weight | H-bond donors | H-bond acceptors | Lipophilicity Log Po/w | Water Solubility (Log S) | Molar Refractivity | GI absorption | BBB permeability | GPCR ligand | Ion channel modulation | Kinase inhibitor | Nuclear receptor ligand | Protease inhibitor | Enzyme inhibitor |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (g/mol) | |||||||||||||||
| Artemisin | 65030 | 262.3 | 1 | 4 | 1.53 | -1.88 | 69.32 | High | Yes | -0.06 | -0.29 | -0.93 | 0.28 | -0.01 | 0.58 |
| Cirsimaritin | 188323 | 314.29 | 2 | 6 | 2.46 | -5.22 | 84.95 | High | NO | -0.09 | -0.24 | 0.2 | 0.17 | -0.31 | 0.14 |
| Curcumin | 969516 | 368.38 | 2 | 6 | 3.03 | -4.45 | 102.8 | High | NO | -0.06 | -0.2 | -0.26 | 0.12 | -0.14 | 0.08 |
| Hispidulin | 5281628 | 300.26 | 3 | 6 | 2.12 | -4.52 | 80.84 | High | NO | -0.07 | -0.22 | 0.21 | 0.2 | -0.33 | 0.17 |
| Quercetin | 5280343 | 302.24 | 5 | 7 | 1.23 | -3.24 | 78.03 | High | NO | -0.06 | -0.19 | 0.28 | 0.36 | -0.25 | 0.28 |
| Sulfasalazine | 5339 | 398.39 | 3 | 8 | 2.3 | -5.86 | 100.95 | Low | NO | 0.03 | -0.21 | -0.02 | -0.38 | 0.05 | 0.09 |
| Thymoquinone | 10281 | 164.2 | 2 | 0 | 1.85 | -2.03 | 47.52 | High | Yes | -1.4 | -0.31 | -1.27 | -1.47 | -1.45 | -0.4 |
| Gallic Acid | 46780424 | 424.44 | 1 | 5 | 4.88 | -9.11 | 119.15 | High | Yes | 0.14 | -0.01 | -0.24 | 0.07 | -0.1 | 0.09 |
| Hydroxy- Chloroquine | 3652 | 335.9 | 2 | 3 | 3.37 | -6.35 | 98.57 | High | Yes | 0.35 | 0.3 | 0.44 | -0.12 | 0.12 | 0.15 |
Table 3: Predicted physiochemical and biological activity of assessed molecules.
Discussions
According to in silico results, Gallic acid, Quercetin, Hispidulin, Cirsimaritin, Sulfasalazine, Artemisin, and Curcuma have a better affinity against COVİD-19 protease better than Hydroxy-Chloroquine. The obtained results show also that Gallic acid, Quercetin, Hispidulin, Cirsimaritin and Sulfasalazine exhibited as the best potential inhibitors against COVID-19 main protease 6LU7. Quercetin is an anti- oxidative flavonoid widely distributed in the plant kingdom, is a dietary antioxidant that prevents oxidation of low-density lipoproteins in vitro [11]. Quercetin is a flavonoid with a wide range of biological activities, is used in many countries as vasoprotectants [12]. Intake of quercetin was inversely associated with coronary heart disease mortality in elderly men [13]. Quercetin displays a large variety of biological activities including anticancer activity [13], cardioprotective [14], antioxidant and antidiabetic effect [15]. The protective effect of quercetin on chloroquine-induced oxidative stress and hepatotoxicity in mice was also approved [16]. According to in silico results, Quercetin have a better affinity against COVİD-19 protease better than Hydroxy- Chloroquine. The obtained results show also that Quercetin exhibited as potential inhibitors against COVID-19 main protease 6LU7. Hispidulin, Cirsimaritin, and Artemisin are the main flavonoids isolated from Artemesia herba alba [17]. Artemesia herba alba displays a large variety of biological activities including antidiabetic, antihyperlipidemic and nephroprotective [18, 19], cardioprotective [20], anticancer [21], antioxidant [19], antiprotozoal [22], gastroprotective [23], antibacterial [24], antihepatotoxic [19], insecticidal [25], Essential oils of Artemesia herba alba have also antihypertensive activities [26].
Sulfasalazine [Salazopyrin®] is an intestinal anti- inflammatory, developed in the 1950s to treat rheumatoid arthritis [27]. According to in silico results, Sulfasalazine have a better affinity against COVİD-19 protease better than Hydroxy-Chloroquine. The obtained results show also that Sulfasalazine exhibited potential inhibitors against COVID-19 main protease 6LU7. Curcuma displays a large variety of biological activities including cardioprotective [28], anticancer [29], antiprotozoal [30], antibacterial [31], antihepatotoxic [32], insecticidal [33], the effect of curcuma in experimental malaria has been also demonstrated by Gomes, et al. [34]. According to in silico results, Curcuma have a better affinity against COVİD-19 protease better than Hydroxy-Chloroquine. The obtained results show also that Curcuma exhibited potential inhibitors against COVID-19 main protease 6LU7. Thymoquinone (TQ) is one of the bioactive component derived from the medicinal plant Nigella sativa. Thymoquinone (TQ) exhibited many biological effects including antihistaminic effect [35], anti-asthmatic [36]. Cardioprotective [36], anticancer [37], antibacterial [38], antihepatotoxic [39], the effect of Thymoquinone in experimental malaria has been also demonstrated by El- Sayed, et al. [40]. According to in silico results, Thymoquinone have a good affinity against COVİD-19 protease but its lower than Hydroxy-Chloroquine. The obtained results show also that Thymoquinone exhibited as potential inhibitors against COVID-19 main protease 6LU7 [41].
Conclusion
Spreading outbreak of COVID-19 has challenged the healthcare sector of the world in the last few months. To contribute to this fight against COVID-19, virtual screening based molecular docking was performed to identify novel compounds having the potential to bind Mpro of COVID-19. Our results demonstrate that Gallic acid, Quercetin, Hispidulin, Cirsimaritin, Sulfasalazine, Artemisin, and Curcuma have a better binding affinity to Mpro of COVID-19 protease better than Hydroxy-Chloroquine. Those molecules can be used as therapeutics against COVID-19. However, further studies should be conducted for the validation of these compounds using in vitro and in vivo models to pave a way for these compounds in drug discovery.
Acknowledgment
The present work was supported by DG-RSDT (General Directorate of Scientific Research and Technological Development-Algeria) via the Environmental Research Center (C.R.E), Campus, Sidi Amar, Annaba 23001; Algeria.
Disclosure Statement: The authors report no conflict of interest.
References
-
Su S, Wong G, Shi W, Liu J, Lai ACK, et al. (2016) Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends in microbiology 24(6): 490-502.
-
Belouzard S, Millet JK, Licitra BN, Whittaker GR (2012) Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4(6): 1011-1033.
-
Mcintosh K, Perlman S (2015) Coronaviruses, including severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS). 8th (Edn.), Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, Philadelphia, PA: Elsevier Saunders.
-
Liu Z, Xiao X, Wei X, Li J, Yang J, et al. (2020) Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS‐ CoV‐2. J Med Virol 92(6): 595-601.
-
Robson B (2020) Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Computers in Biology and Medicine 119: 103670.
-
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, et al. (2020) Network-based drug repurposing for novel oronavirus 2019-nCoV/SARS-CoV-2. Cell Discovery 6: 14.
-
Kitchen D, Decornez H, Furr J, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3(11):935–949.
-
Jin Z, Du X, Xu Y, Deng Y, Liu M, et al. (2020) Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811): 289-293.
-
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, J Comput Chem 31(2): 455-461.
-
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development setting. Advanced Drug Delivery Reviews 46(1-3): 3-26.
-
Hollman PC, de Vries JH, van Leeuwen SD, Mengelers MJ, Katan MB (1995) Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers. The American journal of clinical nutrition 62(6): 1276- 1282.
-
Erlund I, Kosonen T, Alfthan G, Mäenpää J, Perttunen K, et al. (2000) Pharmacokinetics of quercetin from quercetin aglycone and rutin in healthy volunteers. European journal of clinical pharmacology 56(8): 545-553.
-
Murakami A, Ashida H, Terao J (2008) Multitargeted cancer prevention by quercetin. Cancer letters 269(2): 315-325.
-
Li M, Jiang Y, Jing W, Sun B, Miao C, et al. (2013) Quercetin provides greater cardioprotective effect than its glycoside derivative rutin on isoproterenol-induced cardiac fibrosis in the rat. Can J Physiol Pharmacol 91(11): 951-959.
-
Abdelmoaty MA, Ibrahim MA, Ahmed NS, Abdelaziz MA (2010) Confirmatory studies on the antioxidant and antidiabetic effect of quercetin in rats. Indian Journal of Clinical Biochemistry 25(2): 188-192.
-
Kumar Mishra S, Singh P, Rath SK (2013) Protective effect of quercetin on chloroquine-induced oxidative stress and hepatotoxicity in mice. Malaria research and treatment 2013: 141734.
-
Shen XL, Nielsen M, Witt MR, Sterner O, Bergendorff O (1994) Inhibition of [methyl-3H] diazepam binding to rat brain membranes in vitro by dinatin and skrofulein. Zhongguo yao li xue bao= Acta pharmacologica Sinica 15(5): 385-388.
-
Sekiou O, Boumendjel M, Taibi F, Tichati L, Boumendjel A, et al. (2020) Nephroprotective effect of Artemisia herba alba aqueous extract in alloxan-induced diabetic rats. Journal of Traditional and Complementary Medicine.
-
Sekiou O, Boumendjel M, Taibi F, Boumendjel A, Messarah M (2019) Mitigating effects of antioxidant properties of Artemisia herba alba aqueous extract on hyperlipidemia and oxidative damage in alloxan-induced diabetic rats. Archives of physiology and biochemistry 125(2): 163-173.
-
Irshaid F, Mansi K, Bani-Khaled A, Aburjia T (2012) Hepatoprotetive, cardioprotective and nephroprotective actions of essential oil extract of Artemisia sieberi in alloxan induced diabetic rats. Iranian journal of pharmaceutical research: IJPR 11(4): 1227.
-
Khlifi D, Sghaier RM, Amouri S, Laouini D, Hamdi M, et al. (2013) Composition and anti-oxidant, anti-cancer and anti-inflammatory activities of Artemisia herba-alba, Ruta chalpensis L. and Peganum harmala L. Food Chem Toxicol 55: 202-208.
-
Bachrouch O, Ferjani N, Haouel S, Jemâa JMB (2015) Major compounds and insecticidal activities of two Tunisian Artemisia essential oils toward two major coleopteran pests. Industrial Crops and Products 65: 127-133.
-
Laoufi T, Ladj M (2017) Artemisia herba-alba anti- inflammatory activity and gastro-protective effects in mice (Doctoral dissertation, Université de Bouira).
-
Yashphe J, Feuerstein I, Barel S, Segal R (1987) The antibacterial and antispasmodic activity of Artemisia herba alba Asso. II. Examination of essential oils from various chemotypes. International journal of crude drug research 25(2): 89-96.
-
Sharifian I, Hashemi SM, Aghaei M, Alizadeh M (2012) Insecticidal activity of essential oil of Artemisia herba- alba Asso against three stored product beetles. Biharean Biologist 6(2): 90-93.
-
Zeggwagh NA, Farid O, Michel JB, Eddouks M (2008) Cardiovascular effect of Artemisia herba alba aqueous extract in spontaneously hypertensive rats. Methods Find Exp Clin Pharmacol 30(5): 375-381.
-
Taffet SL, Das KM (1983) Sulfasalazine. Digestive diseases and sciences 28(9): 833-842.
-
El-Sayed EM, El-azeem AS, Afify AA, Shabana MH, Ahmed HH (2011) Cardioprotective effects of Curcuma longa L. extracts against doxorubicin-induced cardiotoxicity in rats. J Med Plants Res 5(17): 4049-4058.
-
Chaithongyot S, Asgar A, Senawong G, Yowapuy A, Lattmann E, et al. (2015) Anticancer effects of curcuma C20-dialdehyde against colon and cervical cancer cell lines. Asian Pacific Journal of Cancer Prevention 16(15): 6513-6519.
-
Shahiduzzaman M, Dyachenko V, Khalafalla RE, Desouky AY, Daugschies A (2009) Effects of curcumin on Cryptosporidium parvum in vitro. Parasitology research 105(4): 1155-1161.
-
Kim JE, Kim HE, Hwang JK, Lee HJ, Kwon HK, et al. (2008) Antibacterial characteristics of Curcuma xanthorrhiza extract on Streptococcus mutans biofilm. The Journal of Microbiology 46(2): 228-232.
-
Kiso Y, Suzuki Y, Watanabe N, Oshima Y, Hikino H (1983) Antihepatotoxic principles of Curcuma longa rhizomes. Planta medica 49(11): 185-187.
-
de Souza Tavares W, Akhtar Y, Gonçalves GLP, Zanuncio JC, Isman MB (2016) Turmeric powder and its derivatives from Curcuma longa rhizomes: insecticidal effects on cabbage looper and the role of synergists. Scientific reports 6: 34093.
-
Gomes GS, Maciel TR, Piegas EM, Michels LR, Colomé LM, et al. (2018) Optimization of curcuma oil/quinine- loaded nanocapsules for malaria treatment. AAPS PharmSciTech 19(2): 551-564.
-
Kanter M, Coskun O, Uysal H (2006) The antioxidative and antihistaminic effect of Nigella sativa and its major constituent, thymoquinone on ethanol-induced gastric mucosal damage. Arch toxicol 80(4): 217-224.
-
Gonca E, Kurt Ç (2015) Cardioprotective effect of Thymoquinone: A constituent of Nigella sativa L., against myocardial ischemia/reperfusion injury and ventricular arrhythmias in anaesthetized rats. Pak J Pharm Sci 28(4): 1267-1273.
-
Paramasivam A, Sambantham S, Shabnam J, Raghunandhakumar S, Anandan B (2012) Anti-cancer effects of thymoquinone in mouse neuroblastoma (Neuro-2a) cells through caspase-3 activation with down-regulation of XIAP. Toxicology letters 213(2): 151- 159.
-
Halawani E (2009) Antibacterial activity of thymoquinone and thymohydroquinone of Nigella sativa L. and their interaction with some antibiotics. Advances in Biological Research 3(5-6): 148-152.
-
Daba MH, Abdel-Rahman MS (1998) Hepatoprotective activity of thymoquinone in isolated rat hepatocytes. Toxicology letters 95(1): 23-29.
-
El-Sayed SAES, Rizk MA, Yokoyama N, Igarashi I (2019) Evaluation of the in vitro and in vivo inhibitory effect of thymoquinone on piroplasm parasites. Parasites & vectors 12(1): 37.
-
Keyhanmanesh R, Boskabady MH, Khamneh S, Doostar Y (2010) Effect of thymoquinone on the lung pathology and cytokine levels of ovalbumin-sensitized guinea pigs. Pharmacological reports 62(5): 910-916.
- Superposition of Cryo-EM and AlphaFold Predictions of Dengue Antigen-Antibody Complexes
- Jugular-Applied Coherent Low-Level Laser Therapy Enhances Systemic Mitochondrial Metabolic Function and Antioxidant Response
- Role of OMC32 Polypeptide in Acrosin-Mediated Exocytosis during the Bovine Sperm Acrosome Reaction
- Association of Galectin-3 but not Laminin in Tamoxifen-Induced Growth Suppression in Breast Cancer MCF-7 Cells
- Effect of Different Wavelengths of Light on the Rate of Photosynthesis
- Nutritional, Therapeutic, and Environmental Effect of Oyster Mushrooms: An Editorial