Early Surgical Resection for Symptomatic Congenital Cystic Adenomatoid Malformation (CCAM) in the Newborn Child
Congenital cystic adenomatoid malformation (CCAM) of the lung is a rare congenital abnormality. We present here a one month male child who had no respiratory distress after birth, but gradually developed dyspnea. The diagnosis was made prenatally. After further diagnosis (chest CT wich reveals a large lesion with mediastinal shifting ) due to the deterioration of the clinical picture, surgical treatment is indicated in the first month of life.
Introduction
Congenital cystic adenomatoid malformation (CCAM) of the lung is a rare congenital abnormality. The incidence of CCAM has been reported in approximately 1 out of 25,000 pregnancies [1]. The pathological feature of the lesion is adenomatoid proliferation of bronchioles that form cysts at the expense of normal aleveoli. Its exact pathogenesis is still uncertain although latest literatures state it is due to fetal maldevelopment of the lung bud between the 6th to 8th weeks of gestation [2]. It is diagnosed by prenatal screening at 18-20 weeks of gestation. CCAM has communication between the cystic malformation and the tracheobronal tree, so many of the cysts are filled with air, and blood supply usually receives pulmonary circulation. The wall cyst is made of smooth muscles and elastic tissue, does not contain cartilage. It is limited to a single lobe in 95% of cases and bilateral in less than 2% of cases [3]. CCAM is usually discovered in neonates because of respiratory distress. Eighty percent (80%) are diagnosed before two years of age [4]. The spectrum of this anomaly ranges from asymptomatic lesion to pulmonary compromise. CCAM also may occasionally be discovered in older children or adults who have recurrent infectio.
First classification of this lesion as congenital cystic adenomatoid malformation (CCAM) was given by Chin and Tang in 1949 [5]. Although originally described as “congenital cystic adenomatoid malformation,” this entity renamed as “congenital pulmonary airway malformation” (CPAM) because cystic morphology in not present in all subtypes, nor adenomatoid. Stocker et al. 1977 classified CPAM into three types, based on clinical, macroscopical and microscopical criteria. In 1993 Adzick introduced a classification system. Microcistical lesions (cysts <5 mm) are usually associated with fetal hydrops and have a poor prognosis. Macrocyst lesions (cysts> 5 mm) are not usually associated with hydrops and have a good prognosis [6].
Case report
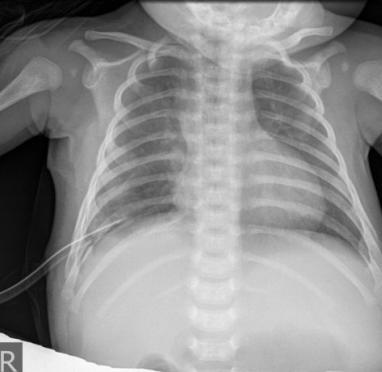
Male infant, terminal, body weight 3440 g and length 57 cm. API = 9/10. There was no need for oxygen support. Prenatal ultrasound examination suspected on congenital diaphragmatic hernia. X ray chest showed right right side with multiple cystic changes with different levels of airborne pulmonary parenchyma (Figure 1).
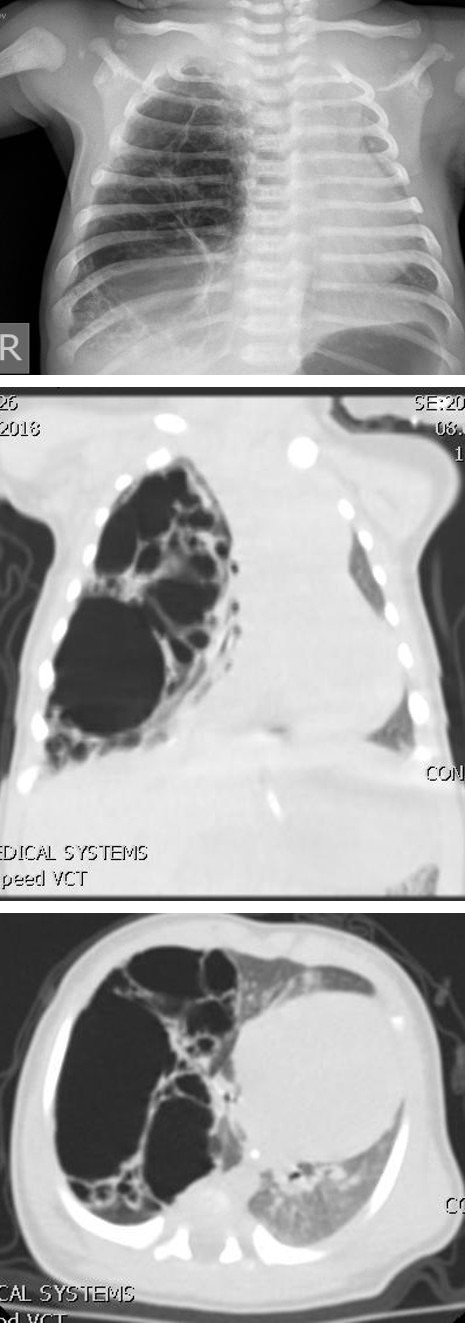
The child is with a normal respiratory status. CT chest is performed: a modified appearance of pulmonary parenchyma in the right half of the thorax. A lumpy parenchyma mostly occupied by hypodontic zones filled with air, thin walls, unrecognizable parenchyma in this area. The cysts are of different sizes from 25 to 61mm. In one of the cysts, noticeable fluency. The cysts are placed in the lower lobe right and compress the pulmonary parenchyma. Cysts suppress mediastinum and large blood vessels on the contralateral left side. No abnormal vascularization of pulmonary parenchyma (Figures 2 & 3).
Figures 2 & 3: The cystic malformation shifting mediastinum on contralateral side.
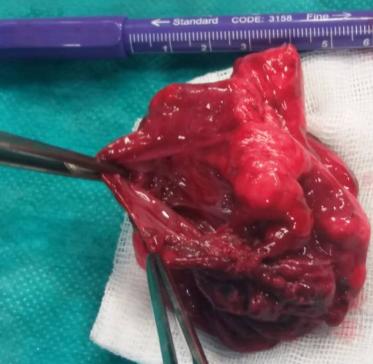
In the course of further observation, the patient becomes dysfunctional and is indicated for surgical treatment due to deterioration of the respiratory status as well as due to the extent of the radiologically described changes. On examination, pallor was present, breath sounds were decreased and percussion note was dull in right region. During the surgery the right thoracotomy is performed and there is a large cystic change taking the lower lobe while the middle and upper pulmonary lobes are atelectical [7]. Typical lower right lobectomy was performed (Figures 4 & 5).
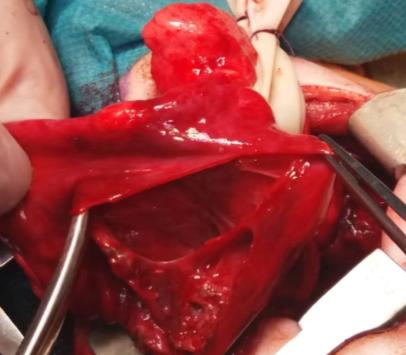
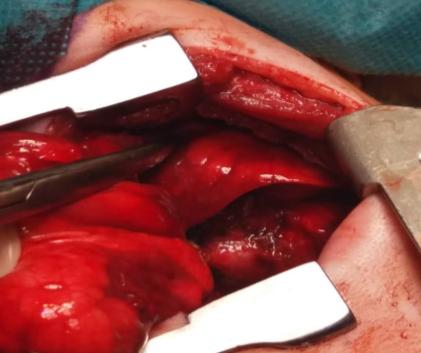
Figures 4 & 5: Right lower lobectomy - right lower lobe cystically changed.
The lower right lobe is removed, completely destroyed by cystic malformation, with very little preserved lung
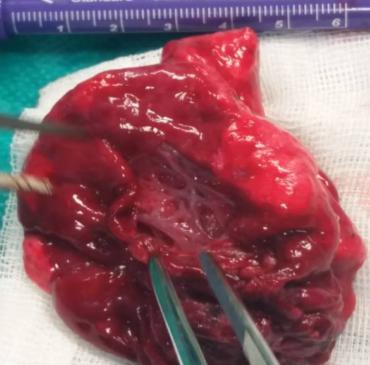
Figures 6 & 7: The lower right lobe is removed, completely destroyed by cystic malformation, with very little preserved lung parenchyma. The postoperative course was satisfactory, antibiotic therapy is ordered. The thoracic tube is removed. The Control Rtg is neat and the patient's postoperative course is satisfactory (Figure 8).
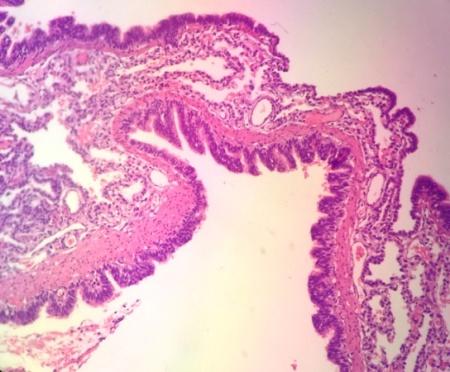
The pathohistological finding has shown a gross appearance of the pulmonary lobe showing several parenchyma (Figures 6 & 7).
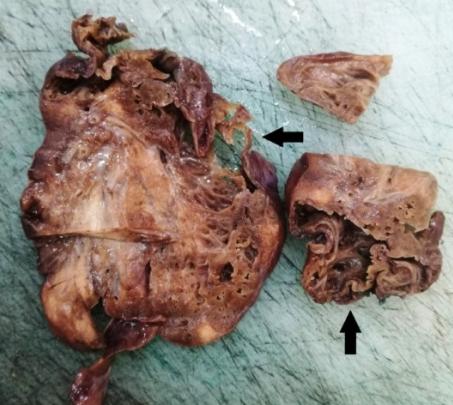
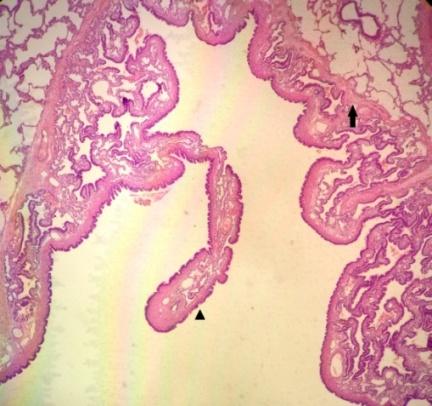
variable-sized cystic formations (larger than 2 cm) outlined by thin walls with honeycomb appearance (Figures 9-11).
The patient is released from the hospital at home treatment.
Discussion
CCAM is a very rare lung abnormality but the most common congenital cystic lung diseases. CCAM most commonly present with respiratory distress due to the expanding cystic lung mass while others are asymptomatic and later discovered on chest radiograph. Cough, fever, and repeated respiratory infections are the less common presentations. Routine prenatal US screening has become increasingly valuable in detecting CCAM [8] with an accuracy of 65–91% [9, 10]. Chest X-ray, computed tomography (CT) scan and clinical follow-up after birth is required in all cases to confirm the diagnosis and to initiate adequate treatment [11, 12]. Significant advances in neonatal intensive care have allowed support, either with conventional ventilation or high-frequency oscillation ventilation, optimising oxygen delivery. A postnatal echocardiogram is essential for assessing cardiovascular function.
Emergency operation for CCAM is performed when the patient has symptoms of respiratory distress in the neonatal period and subsequent diagnostic work-up reveals CCAM. Some newborns with large lesions even asymptomatic, may require a neonatal surgical resection in order to save viable lung parenchyma and reverse mediastinal displacement [7]. In asymptomatic patients, the diagnosis of CCAM is not usually made until childhood or later, when symptoms of frequent pneumonia develop or an incidentally checked chest roentgenography reveals the lesion. In those patients, elective surgical resection is recommended at the time of diagnosis. There is no clear indication for deciding the best timing for operation when the diagnosis is made in utero by fetal USG.
The development of neonatal care, thoracic surgical techniques, anesthesiology, nursing care, and postoperative management all enable safe surgery, even for a neonate. Many reports recommend early surgery for infants in all cases of CCAM [13]. Our opinion that the respiratory distress syndrome in patients with CCAM is sufficient reason for emergency operation. If the patient is asymptomatic and the CT scan shows mediastinal shifting and large cystic lesion, then urgent surgery is required. Aziz and al. suggested that the non-operative approach might be a reasonable alternative for asymptomatic children with perinatally diagnosed CCAM because there was no increase in the postoperative complication rate even if surgery was delayed until the onset of symptoms [14]. Another problem of conservative follow up is the risk of malignancy in which true incidence is still unclear. Our opinion is that an asymptomatic patient with CCAM should be clinically monitored and operated after the first year of life and without symptoms of the respiratory tract to prevent complications such as lung infections, pneumothorax and questioneble malignancy Young Tae Kim at al. decided to perform an operation if the patient showed any signs of respiratory distress or there was any evidence of the lesion progressing to develop symptoms. Among five emergency cases which thay had, only one patient suffered dyspnea and the remaining four were operated on an urgent basis as the lesion was too big or had increased in size, even though patients exhibited no symptoms [15].
The extent of surgical resection is also controversial, but since most lesions are limited to the lobe, lobectomy is the mainstay surgical method. A more conservative policy of segmentectomy, when possible, seems to have been employed by some investigators [16, 17, 18]. Lobectomy is the procedure of choice for patients with CCAM, especially when the lesion is confined to a single lobe. Infants and children tolerate lobectomy well, with compensatory lung growth, so that the total lung volume and gas exchange capacity return toward normal during somatic maturation [19, 20]. Our opinion is that this conservative surgery has several disadvantages, such as persistent air-leak or incomplete excision of the lesion. That is why we always apply lobectomy. Only when multiple lobes are affected in patients with CCAM, then extensive resection shoud be avoid and performe parenchyma-saving procedure.
Conclusions
-CCAM neonatal patient with signs of respiratory distress, need a emergency operation. -CCAM neonatal asymptomatic patient with the chest CT wich reveals a large lesion with mediastinal shifting need urgent operation. - Lobectomy is the procedure of choice in neonatal patients with CCAM
References
-
Kanavi JV, Thangaraj J, Thomas A (2017) Congenital cystic adenomatoid malformation (CCAM): antenatal and postnatal management Int J Reprod Contracept Obstet Gynecol. 6(6): 2447-2451.
-
Singh J (2011) Congenital Cystic Adenomatous Malformation: A Case Report. JIMSA 24(2).
-
Carias MJB, Orillaza M (2007) Congenital Cystic Adenomatoid Malformation Type II with Associated Cardiac Anomalies. Accepted paper for PHC 15th Annual research Paper Competition 2007 and for 38th PHA Annual Convention held on May 16-18, 2007 at Edsa Shangrila Hotel, Philippines 13(2): 161- 167.
-
David M, Lamas-Pinheiro R, Henriques-Coelho T (2016) Prenatal and Postnatal Management of Congenital Pulmonary Airway Malformation. Neonatology 110(2): 101-115.
-
Adzick NS (2003) Management of fetal lung lesions. Clin perinatol 30(3): 481-492.
-
Vu L, TsaoK, Lee M, Nobuhara K, Farmer D, et al. (2007) Characteristics of congenital cystic adenomatoid malformations associated with non immune hydrops and outcome. J Pediatr Surg 42(8): 1351-1356.
-
Kotecha S, Barbato A, Bush A, Claus F, Davenport M, et al. (2012) Antenatal and postnatal management of congenital cystic adenomatoid malformation. Paediatr Respir Rev 13(3): 162-171.
-
Breysem L, Bosmans H, Dymarkowski S, Schoubroeck DV, Witters I, et al. (2003) The value of fast MR imaging as an adjunct to ultrasound in prenatal diagnosis. Eur Radiol 13(7): 1538-1548.
-
Tsai AY, Liechty KW, Hedrick HL, Bebbington M, Wilson RD, et al. (2008) Outcomes after postnatal resection of prenatally diagnosed asymptomatic cystic lung lesions. J Pediatr Surg 43(3): 513-517.
-
Farrugia MK, Raza SA, Gould S, Lakhoo K (2008) Congenital lung lesions: classification and concordance of radiological appearance and surgical pathology. Pediatr Surg Int 24(9): 987-991.
-
Raychaudhuri P, Pasupati A, James A, Whitehead B, Kumar R (2011) Prospective study of antenatally diagnosed congenital cystic adenomatoid malformations. Pediatr Surg Int 27(11): 1159-1164.
-
Ankermann T, Oppermann HC, Engler S,Leuschner I, von Kaisenberg CS (2004) Congenital masses of the lung, cystic adenomatoid malformationversus congenital lobar emphysema.J Ultrasound Med 23(10): 1379-1384.
-
Miller JA, Corteville JE, Langer JC (1996) Congenital cystic adenomatoid malformation in the fetus: natural history and predictors of outcome.J Pediatr Surg 31(6): 805-808.
-
Aziz D, Langer JC, Tuuha SE, Ryan G, Ein SH, et al. (2004) Perinatally diagnosed asymptomatic congenital cystic adenomatoid malformation:to resect or not? J Pediatr Surg 39(3): 329-334.
-
Kim YT, Kim JS, Park JD, Kang CH, Sung SW, et al. (2005) Treatment of congenital cystic adenomatoid malformation—does resection in the early postnatal period increase surgical risk? European Journal of Cardio-thoracic Surgery 27 (4): 658-661.
-
Sapin E, Lejeune V, Barbet JP, Carricaburu E, Lewin F, et al. (1997) Congenital adenomatoid disease of the lung: prenatal diagnosis and perinatal management. Pediatr Surg Int 12(2-3): 126-129.
-
Waszak P, Claris O, Lapillonne A, Picaud JC, Basson E, et al. (1999) Cystic adenomatoid malformation of the lung: neonatal management of 21 cases. Pediatr Surg Int 15(5-6): 326-331.
-
Kim HW, Choi YS, Kim K, Shim YM, Ku GW, et al. (2008) Treatment of Congenital Cystic Adenomatoid Malformation: Should Lobectomy Always Be Performed? Ann Thorac Surg 86(1): 249-253.
-
Sauvat F, Michel J, Benachi A, Emond S, Revillon Y(2003) Management of asymptomatic neonatal cystic adenomatoid malformations. J Pediatr Surg 38(4): 548-552.
-
Zach MS, Eber E (2001) Adult outcome of congenital lower respiratory tract malformations. Thorax 56(1): 65-72.
- Management of Chronic Insertional Achilles Tendinopathy Using Flexor Hallucis Longus Tendon Transfer in Patients Over 50 Years of Age: A Four-Case Series Following the CARE Guidelines
- Application of Induced Pluripotent Stem Cells in Bone Tissue Engineering: Current Status and Prospects
- Surgical Management of Upper Thoracic Esophageal Squamous Cell Carcinoma with Concomitant Hypersplenism: Integration of Chai's Supra-Thoracic Apex Technique with Laparoscopic Splenectomy - A Technical Innovation Case Study with Systematic Review
- Evaluation of Masticatory Functional Efficiency of Stomatognathic System in Patients Undergoing Open Reduction Internal Fixation for Treatment of Pan-Facial Trauma: A Prospective Study
- Hepatic Abscess Secondary to Appendiceal Phlegmon an Unusual Complication of Appendiceal Phlegmon
- Report of Lumboperitoneal (LP) Shunt Procedure in Over Decades Experiences, Systematic Narrative Review