Vacterl Association with Mayer-Rokitansky-Kuster-Hauser Syndrome, Description of a Case
Vacterl association is a complex dysfunction of congenital malformaciones that implies several structures. Is typically defined by the presence of at least three of the following congenital malformations: vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal anomalies, and limb abnormalities. Etiology has only been identified in some patients due to sporadic nature and her high degree of clinical heterogeneity. We report a girl with VACTERL association, which co-occurrence with unusual anomalies of the branchial arch: Mayer-Rokitansky-Küster-Hause syndrome (MRKHS). Vaginal agenesis, is a clinical malformations due to severe dysfunctions in the development of Muller conduits in feminine patient, with absence of vagina and uterus, but with functionals ovaries. Therapeutic management is typically multidisciplinary and centers around surgical correction of specific congenital anomalies in the immediate postnatal period, followed by long-term follow-up for postoperative complications and sequelae of congenital malformations. If optimal surgical correction is achievable, the prognosis may be relatively positive, although some patients will remain affected throughout life. This presentation constitutes a new modality of presentation of polyformative syndromes that need of experience and medical investigation for patients with complex malformation to improve a quality of life.
Introduction
The complex VACTERL association described in 1973 by Quan and Smith, is an association of congenital malformations with several anomalies that imply vertebraes, anus, heart, trachea, esophagus, kidneys and extremities [1].
This sporadic findings are conceptualized as non aleatory associations that presented in at least two individuals, which happen at the same time with more frequency.
Mayer-Rokitansky-Küster-Hause (MRKHS) syndrome is a congenital malformation with severe dysfunctions in the development of Müller conduits. This name appear in honor to diferents contributions: Karl Mayer AFJ, et al. [2].
The co-occurrence of the VACTERL association and MRKH syndrome is extremely rare and has only been casuistically reported. Even with optimal surgical corrections of malformations, patients affected by the VACTERL association can face medical challenges throughout life such as back pain (scoliosis), fecal incontinence (anal atresia, AA), and functional impairment (limb anomalies) [3].
Reporting cases of rare conditions is important to expand knowledge on treatment, and outcome. The medical challenges are highly dependent on type and severity of the specific malformation. We have treated one patient at our institution with the VACTERL association and MRKH co- occurrence.
Case Presentation
A girl was delivered by caesarean section at 39 weeks of gestation. Her birthweight was 2600 g and her Apgar scores were 8/1 and 9/5. Parent´s wasn’t refer exposure teratogenic agents.
Diagnostic Evaluation
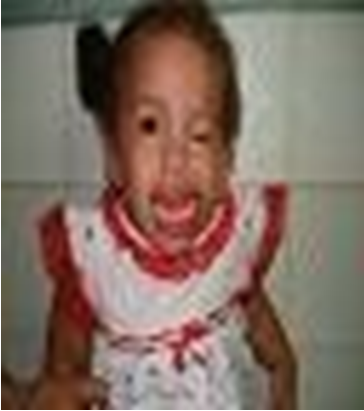
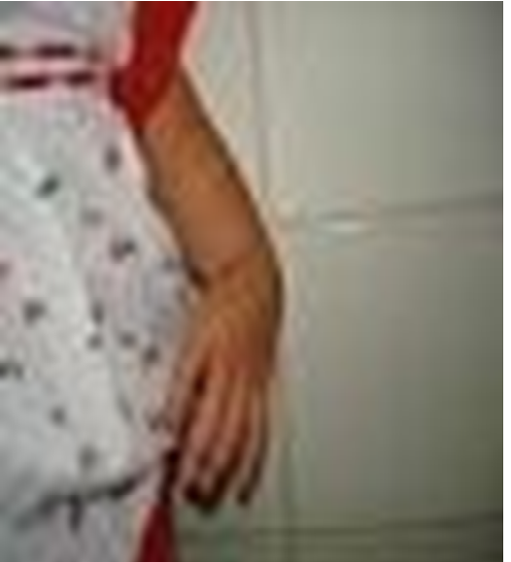
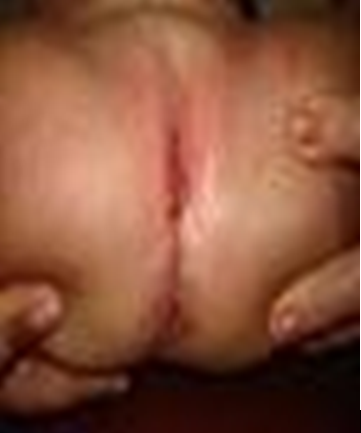
A clinic evaluation and physical examination reveals: equine varus foot, facial dysmorphia, oral cavity with high palate, preauricular apendix, low set ear, right hand polydactyly, absence of left radius and thumb, extreme short neck, left microphthalmia, tracheoesophageal fistula, anterior anus located more anteriorly along perineal body with normal caliber, lumbar thoracic deformity absence of sacrum, vaginal and uterus agenesis, left renal ectopia and ventricular communication (Figures 1-5).
The chromosomal analysis of the anniotic fluid confirmed a normal female karyotype (46XX).


Therapeutic Intervention
Plain X-ray indicated esophageal atresia with TEF. End- to-end anastomosis of her oesophagus and closure of the TEF was perfomed when she was 36 hours with good evolution (Figure 6).
Subsequently was made repair equinus varus foot with later surgical intervations (Figure7).

Laparoscopy exploration was perfomed for diagnostic purposes and the major abdominal cavity finding reveals: persistence, of gonads remainders with fibrous cord. Biopsies inform normal feminine Karyotype, 46 XX.
A diverging colostomy was created, despite a presence anterior anus located more anteriorly along perineal body to avoid infection and future functional alterations. Posterior sagittal anorectoplasty (PSARP) with vaginoplasty with ileum was performed when she was 3 months of age. Colostomy closure was made 6 months later (Figures 8).
Follow Up and Results
A renography performed at 1 year of age showed normal function of her left kidney. Non alterations of the central nervous system with adapted audition and understanding, single slight difficulty of the language. At the moment patient continues alive with appropriate locomotion, swallowing and normal defecations with continence.
Discussion
The co-occurrence of the VACTERL association and MRKH syndrome is extremely rare and we have only detected nine reported cases but added one from our country.
VACTERL association is typically defined by the presence of at least three of the following congenital malformations: vertebral defects, anal atresia, cardiac defects, tracheo- esophageal fistula, renal anomalies, and limb abnormalities. The prevalence has been estimated to be 1:10,000 to 1:40,000 in live births [4].
The etiology remains largely unknown, probably due to its clinical and causal heterogeneity, the typical sporadic nature of the disease, and the many overlapping conditions [5].
Vertebral anomalies have been reported in 60 to 80% of patients and characteristic vertebral anomalies include segmentation defects such as hemivertebrae, vertebral fusions, supernumerary or absent vertebrae, and dysplastic butterfly-shaped or wedge- shaped vertebrae. They can commonly be accompanied by rib anomalies, but these can appear alone without vertebral changes [6].
They are also associated with abnormal spinal curvatures, due to the coexistence of underlying costovertebral anomalies. In this sense, the appearance of clinical signs of scoliosis may be the first sign when the association with VACTERL is suspected.
Anorectal malformation is reported in 55 to 90% of patients with VACTERL association. In contrast to vertebral anomalies, anorectal malformation is often recognized during the immediate clinical examination after birth. In more subtle cases anorectal malformation may present later as constipation, which appears in up to 21% of the patients.
Some clinicians require an imperforate anus to address it as a VACTERL association while others allow minor anorectal malformation to be included [7].
The recommended surgical technique for repair of anorectal malformations with rectovestibular fistula is a PSARP, which is reported to have an excellent functional outcome in highly specialized centers and no associated defects [8].
Cardiac malformations are also common and observed in 40 to 80% of patients and including vascular anomalies, that may not be clinically significant to complex malformations necessitating multiple surgical and longterm medical intervention [9].
They range from life threatening malformations, which require complicated surgical procedures, to small asymptomatic defects which are discovered incidentally. A persistent ductus arteriosus and foramen ovale should not be registered as a defect in the VACTERL association [10].
The identification of limb anomalies has been reported in 40–55% of affected individuals and can help plan interventions, including early physical therapy and eventual surgery.
The management of patients with VACTERL association can be complex, so the nuances must be individualized when treating problems related to each characteristic of the component.
In general, the therapeutic management of affected patients can be divided into two fundamental stages. First, conditions that are incompatible with life are treated, such as severe cardiac malformations, anorectal malformations, and tracheoesophageal fistula, which generally require surgery in the immediate neonatal period or as soon as circumstances permit.
Secondly, there are many of the associated congenital malformations, which can cause longer-term sequelae, which is why they require multidisciplinary evaluation and follow-up, to avoid the repercussions that these anomalies, and postoperative complications in particular, may affect the quality of life of these patients.
In the case of anorectal malformation, the timing and surgical approach can differ drastically depending on the clinical presentation, the anatomical variety and the presence of associated malformations, mainly those of the genitourinary system. A staged treatment is generally performed in which an immediate diverting colostomy is initially performed, followed later by a posterior sagittal anorectoplasty [11].
Tracheoesophageal fistula usually requires surgery in the first few days of life and is typically repaired in a single intervention, although later complications such as fistula recurrence, reactive airway disease, and gastroesophageal reflux may require further procedures [12, 13].
Congenital costovertebral anomalies may require orthopedic follow-up and in some cases require surgical interventions, the repair of which is indicated at the earliest ages possible, in relation to the specific procedure to be performed. Early detection of the tethered spinal cord allows for timely surgery, which has shown significant beneficial results, although the benefit may differ in symptomatic patients compared to asymptomatic patients [14].
Early diagnosis and management can preserve function through infection prevention and management of vesicoureteral reflux (VUR). Adequate optimization of bladder emptying is especially important in obstructive hydronephrosis. Eventually a kidney transplant may be necessary, in those serious anomalies that compromise kidney function [15].
In the case of limb anomalies, their identification can help in planning interventions, rehabilitation therapy and eventual surgery in patients who need it [16].
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, also known as Müllerian aplasia, is a disorder characterized by agenesis of the uterus and upper vagina in women with normal secondary sexual characteristics and a normal female karyotype (46XX). It has an estimated prevalence of 1 in 5,000 live births [17].
The exact etiology is still unknown, due to the complexity of the genetic pathways involved during the embryogenetic development of the Müllerian ducts. It is classified as type I isolated uterovaginal aplasia or associated with type II extragenital manifestations. Extragenital abnormalities often include renal, skeletal, ear, or cardiac malformations [18, 19].
Diagnosis is often made during adolescence after investigations for primary amenorrhea. Ultrasound and magnetic resonance imaging are the most widely used fundamental diagnostic means [20, 21].
The diagnosis and work-up in these patients has become very efficient, thanks to the use of imaging, and there are multiple successful procedures for the creation of a neovagina. The treatment of vaginal agenesis is based on successful procedures for the creation of aneovagina, with non-invasive vaginal dilations, recommended as first-line therapy or by surgery [22, 23, 24, 25, 26, 27, 28].
This syndrome implies absolute uterine factor infertility, whose main therapeutic options are in vitro fertilization using autologous oocytes and a surrogate gestational carrier, as well as uterine transplantation to achieve biological motherhood [29, 30].
This syndrome can have a great psychological and/or psychosexual impact and requires continuous psychological counseling and therapeutic education [31, 32].
Conclusion
VACTERL association and MRKHS syndromes can be considered strange co- occurrence which don’t find any national case reported. Therapeutic management is typically multidisciplinary and centers around surgical correction of specific congenital anomalies in the immediate postnatal period, followed by long-term follow-up for postoperative complications and sequelae of congenital malformations. If optimal surgical correction is achievable, the prognosis may be relatively positive, although some patients will remain affected throughout life. Constitutes a new modality of presentation of polyformative syndromes that need experience and medical investigation for patients with complex malformation to improve a quality of life.
Author Contributions
Conceptualization, W.Q.N; methodology, W.Q.N; validation, W.Q.N; formal analysis, W.Q.N; writing—original draft preparation, W.Q.N; writing— review and editing, W.Q.N; visualization, W.Q.N; supervision, W.Q.N. All authors have read and agreed to the published version of the manuscript.
Ethical Considerations
The authors declare that they have followed the protocols and guidelines of their work center, maintaining the confidentiality of the patient’s data. In the same way, informed consent has been obtained from the legal representative of the patient for the publication of the clinical case.
Conflict of Interests
The authors declare that they have no conflict of interest.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets used and analyzed in the current study are available from the corresponding author on demand.
References
-
Solomon BD, Baker LA, Bear KA, Cunningham BK, Giampietro PF, et al. (2014) An approach to the identification of anomalies and etiologies in neonates with identified or suspected VACTERL (vertebral defects, anal atresia, tracheo-esophageal fistula with esophageal atresia, cardiac anomalies, renal anomalies, and limb anomalies) association. J Pediatr 164(3): 451-457.
-
Chen N, Song S, Bao X, Zhu L (2022) Update on Mayer- Rokitansky-Küster-Hauser syndrome. Front Med 16(6): 859-872.
-
Bjørsum Meyer T, Herlin M, Qvist N, Petersen MB (2016) Vertebral defect, anal atresia, cardiac defect, tracheoesophageal fistula/esophageal atresia, renal defect, and limb defect association with Mayer- Rokitansky-Küster-Hauser syndrome in co-occurrence: two case reports and a review of the literature. J Med Case Rep 10(1): 374.
-
National Organization for Rare Disorders (2015) VACTERL Association.
-
Solomon BD (2018) The etiology of VACTERL association: Current knowledge and hypotheses. American Journal of Medical genetics 178(4): 440-446.
-
Orphanet Report Series (2022) Prevalence and incidence of rare diseases: Bibliographic data.
-
Pena A, Bischoff A (2015) Rectovestibular fistula. In: Pena A, Bischoff A, (Eds.), Surgical treatment of colorectal problems in children pp: 205-224.
-
Teo XL, Narasimhan KL, Chua JHY (2015) Mullerian agenesis in the presence of anorectal malformations in female newborns: a diagnostic challenge. Singapore Med J 56(5): 82-84.
-
Avila Iglesias MC, Rojas Maruri CM (2017) Asociación VACTERL. Presentación de un caso en sesión anatomo- patológica y consideraciones generales. Acta pediatr. Méx 38(5): 330-336.
-
Chin C (2013) ABCs of Neonatal Cardiac Anesthesia. Artif Organs 37(1): 100-102.
-
Lautz TB, Mandelia A, Radhakrishnan J (2015) VACTERL associations in children undergoing surgery for esophageal atresia and anorectal malformations: Implications for pediatric surgeons. J Pediatr Surg 50(8): 1245-1250.
-
Morgan RD, O’Callaghan JM, Wagener S, Grant HW, Lakhoo K (2012) Surgical correction of tracheooesophageal fistula and oesophageal atresia in infants with VACTERL association: a retrospective case-control study. Pediatr Surg Int 28(10): 967-970.
-
Yang L, Li S, Zhong L, Qiu L, Xie L, et al. (2019) VACTERL association complicated with multiple airway abnormalities: A case report. Medicine 98(42): e17413.
-
Contreras Omaña R, Aguilar Lira JL (2014) VACTERL syndrome. Rev Gastroenterol Mex 79(2): 147-148.
-
Telkes G, Reusz G, Szabo AJ, Langer RM (2011) A single- center experience with kidney transplantation in the verteberal, anal, cardiac, tracheoesophageal, renal, and limb birth detects (VACTERL) association. Transplant Proc 43: 1250-1251.
-
Oral A, Caner I, Yigiter M, Kantarci M, Olgun H, et al. (2012) Clinical characteristics of neonates with VACTERL association. Pediatr Int 54(3):361-364.
-
Herlin M, Bjorn AMB, Rasmussen M, Trolle B, Petersen MB (2016) Prevalence and patient characteristics of Mayer-Rokitansky-Küster-Hauser syndrome: a nationwide registry-based study. Hum Reprod 31(10): 2384-2390.
-
Backhouse B, Hanna C, Robevska G, van den Bergen J, Pelosi E, et al. (2019) Identification of candidate genes for Mayer-Rokitansky-Kuster-Hauser syndrome using genomic approaches. Sex Dev 13(1): 26-34.
-
Deng S, He Y, Chen N, Zhu L (2019) Spectrum of type I and type II syndromes and associated malformations in Chinese patients with Mayer-Rokitansky- KusterHauser syndrome: a retrospective analysis of 274 cases. J Pediatr Adolesc Gynecol 32(3): 284-287.
-
Cekdemir YE, Mutlu U, Acar D, Altay C, Secil M, et al. (2022) The accuracy of three-dimensional ultrasonography in the diagnosis of Müllerian duct anomalies and its concordance with magnetic resonance imaging. J Obstet Gynaecol 42(1): 67-73.
-
Kim DY, Nam G, Lee SR, Kim SH, Chae HD, et al. (2021) Congenital Obstructive Mullerian Anomaly: The Pitfalls of a Magnetic Resonance Imaging- Based Diagnosis and the Importance of Intraoperative Biopsy. J Clin Med 10(11): 2414.
-
Kapczuk K, Iwaniec K, Friebe Z, Kedzia W (2016) Congenital malformations and other comorbidities in 125 women with Mayer-Rokitansky-Kuster-Hauser syndrome. Eur J Obstet Gynecol Reprod Biol 207: 45-49.
-
Berglund A, Burt E, Cameron-Pimblett A, Davies MC, Conway GS (2019) A critical assessment of case reports describing absent uterus in subjects with oestrogen deficiency. Clin Endocrinol 90(6): 822–826.
-
Herlin MK, PetersenMB, Brännström M (2020) Mayer- Rokitansky-Küster- Hauser (MRKH) syndrome: a comprehensive update. Orphanet J Rare Dis 15(1): 214.
-
American College of Obstetricians and Gynecologists Committee Opinion (2018) Mullerian Agenesis: Diagnosis, Management, and Treatment. Obstet Gynecol 131: e35-e42.
-
Ding JX, Chen L, Zhang X, Zhang Y, Hua KQ (2015) Sexual and functional outcomes of vaginoplasty using acellular porcine small intestinal submucosa graft or laparoscopic peritoneal vaginoplasty: a comparative study. Hum Reprod 30(3): 581-589.
-
Cheikhelard A, Bidet M, Baptiste A, Viaud M, Fagot C, et al. (2018) Surgery is not superior to dilation for the management of vaginal agenesis in Mayer Rokitansky- Kuster-Hauser syndrome: a multicenter comparative observational study in 131 patients. Am J Obstet Gynecol 219(3): 281e1-281e9.
-
Zhang X, Liu Z, Yang Y, Yao Y, Tao Y (2017) The clinical outcomes of vaginoplasty using tissue-engineered biomaterial mesh in patients with Mayer Rokitansky- Küster-Hauser syndrome. Int J Surg 44: 9-14.
-
Brannstrom M, Bokstrom H, Dahm Kahler P, Diaz-Garcia C, Ekberg J, et al. (2016) One uterus bridging three generations: first live birth after motherto- daughter uterus transplantation. Fertil Steril 106(2): 261-266.
-
Molne J, Broecker V, Ekberg J, Nilsson O, Dahm-Kahler P, et al. (2017) Monitoring of human uterus transplantation with cervical biopsies: a provisional scoring system for rejection. Am J Transplant. 17(6): 1628-1636.
-
Kang J, Chen N, Song S, Zhang Y, Ma C, et al. (2020) Sexual function and quality of life after the creation of a neovagina in women with Mayer Rokitansky-Küster- Hauser syndrome: comparison of vaginal dilation and surgical procedures. Fertil Steril 113(5): 1024-1031.
-
Pastor Z, Fronek J, Novackova M, Chmel R (2017) Sexual life of women with Mayer- Rokitansky-Kuster-Hauser syndrome after laparoscopic Vecchietti Vaginoplasty. Sex Med 5(2): e106-e113.
- Management of Chronic Insertional Achilles Tendinopathy Using Flexor Hallucis Longus Tendon Transfer in Patients Over 50 Years of Age: A Four-Case Series Following the CARE Guidelines
- Application of Induced Pluripotent Stem Cells in Bone Tissue Engineering: Current Status and Prospects
- Surgical Management of Upper Thoracic Esophageal Squamous Cell Carcinoma with Concomitant Hypersplenism: Integration of Chai's Supra-Thoracic Apex Technique with Laparoscopic Splenectomy - A Technical Innovation Case Study with Systematic Review
- Evaluation of Masticatory Functional Efficiency of Stomatognathic System in Patients Undergoing Open Reduction Internal Fixation for Treatment of Pan-Facial Trauma: A Prospective Study
- Hepatic Abscess Secondary to Appendiceal Phlegmon an Unusual Complication of Appendiceal Phlegmon
- Report of Lumboperitoneal (LP) Shunt Procedure in Over Decades Experiences, Systematic Narrative Review