Review: Treacher Collins Syndrome: A Brief Review on Diagnostic Aids and Review of Literature with a Case Description
Importance: Treacher Collins syndrome (TCS), also called Treacher Collins-Franceschetti syndrome or mandibulofacial dysostosis, is an autosomal dominant disorder. This condition affects an estimated 1:10,000 to 1:50,000 of living births. Observations: This paper describes a 19 year old male patient suspected with Treacher Collins syndrome. He displayed a moderate form of the disease and a classic phenotype, demonstrating many cardinal features of the disorder. Conclusion and Relevance: Along with diagnostic aids which are helpful in diagnosing the disease, this paper also reviews the literature and syndrome in the light of recent publications with emphasis on pathogenesis (role of neural crest cells) and broad discussion on the syndrome as a whole.
Introduction
Treacher Collins syndrome (TCS), also called Treacher Collins-Franceschetti syndrome or mandibulofacial dys- ostosis, is an autosomal dominant disorder. The development of structures derived from the first and second brachial arches during early embryonic development is affected by this syndrome [1].
The syndrome is named after Edward Treacher Collins (1862–1932), who was an English surgeon and ophthalmologist, who described the syndrome in 1900. The gene which carries the genetic alteration has been mapped in the distal portion of the chromosome 5 long arm (5q31.3‑q33.3) [2].
Conceptual Paper
To date, TCS has been proven to be genetically heterogeneous, and three genes have been identified in the aetiology of TCS; however, the complete aetiology of TCS is still not fully elaborated. Pathogenic mutations in about 11% of TCS cases in the population remain unknown, suggesting that there might be other disease- related genes involved in TCS. Furthermore, the relationship between the genotype and phenotype is not fully understood. The known causative genes for TCS are TCOF1, POLR1D and POLR1C. TCOF1 is responsible for approximately 70–93% of studied individuals with autosomal dominant cases. To date, more than 200 distinct mutations have been identified within the TCOF1 gene and most of the mutations are family- specific (The Human Gene Mutation Database®). POLR1D and POLR1C mutations account for 11–23% of patients without TCOF1 mutations [3].
This condition affects an estimated 1:10,000 to 1:50,000 [4] of living births. The signs and symptoms of this disorder vary greatly, ranging from almost unnoticeable to severe [5].
The characteristics 0f TCS are deafness, hypoplastic facial bones namely mandible, maxilla and cheek bone, antimongoloid slant of palpebral fissures, coloboma of lower lid and auricular anomalies. It is a condition in which the cheek bones and jawbones are underdeveloped [6]. TCS patients exhibits a wide spectrum of features ranging from obstruction of perinatal airway leading to death, to mild features. The typical physical features include, as described above, downward‑slanting eyes, micrognathia (a small lower jaw), hearing loss, underdeveloped zygoma, underdeveloped mandibular condyles and temporomandibular joints, drooping part of the lateral lower eyelids, and malformed or absent ears [7, 8, 9].
Case Report
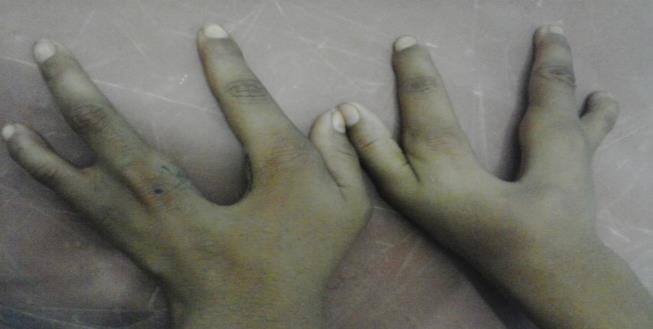
A 19-year-old male reported to the outpatient department of Plastic Surgery, Hamdard institute of Medical Sciences and Research, New Delhi, India; complaining of asymmetry of his face, illformed pinna on right side and deafness (Figures 1a1b). Although short stature and low weight is not typical characteristics of TCS, the patient was 135 cm tall, weight was 38 kg and pallor was observed. On both the upper limbs, the middle fingers were missing and both lower limbs were normal (Figure 2).
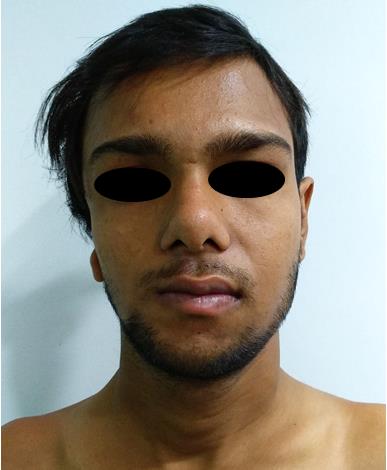
Figure 1a: Frontal view of patient with asymmetry of his face, illformed pinna on right side.
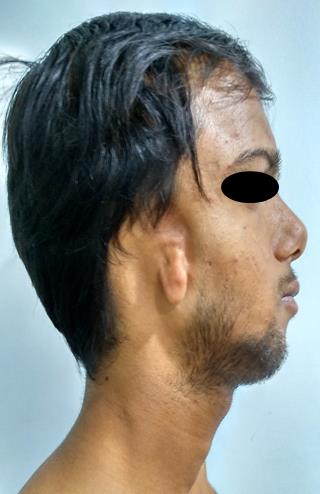
Figure 1b: lateral view of the patient with absent external auditory meatus, microtia, hypoplastic mandibular body on right side.
Extraoral Findings
Ophthalmological findings: On examination, the fundus was bilaterally hypermetropic with macular dystrophy.
ENT findings: Conductive hearing loss, small illformed pinna on right side, absent external auditory meatus on right side, narrow right nasal cavity, soft palate drooping downward.
Facial dysmorphic features: Microcephaly, micrognathia, hypoplastic mandible on right side, and deviated tongue towards right side.
Intraoral Findings
High-arched palate, malocclusion, mouth breathing, open bite, micrognathia. Cleft palate was however absent in our patient.
Investigations
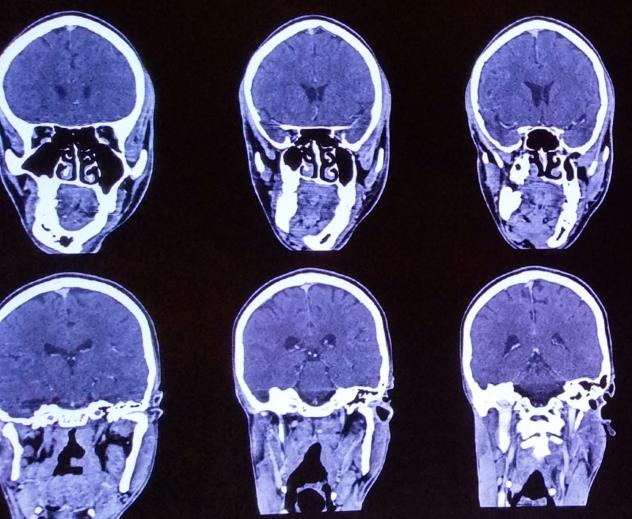
Orthopantomograph, lateral cephalogram, CT, blood investigations (Figure 3).
Diagnosis
Based on the history and clinical features and ra- diographic examination, the patient was diagnosed as having TCS.
Pathogenesis
Abnormal neural crest migration, ectopic cell death and inappropriate differentiation have all been hypothesized as underlying causes of TCS. This syndrome is primarily a neural crest (NC) defect. NC cells contribute to a majority of face and display stem cell features, capable of differentiating into many cell types: neurons, muscle cells, bone, cartilage etc. [10, 11]. Neural crest cells migrate over extensive distances to the periphery of the face giving rise to most of the cartilage, bone, connective and peripheral tissues in the head. Most disorders of craniofacial development are thought to be caused by defects in the formation, proliferation, migration and/or differentiation of cranial neural crest cells and TCS is no exception.
Diagnostic Clinical Features
These are the diagnostic features of TCS: I. Ears
a. Anomalies of the pinna b. External auditory canal abnormalities c. Bony deformity of middle ear cavity d. Variable degrees conductive hearing loss results from hypoplasia of the external auditory canals and ossicular abnormality of the middle ears. II. Eyes a. Antimongoloid slant of the palpebral fissures b. Colobomata and hypoplasia of the lower lids and lateral canthi c. Partial absence of eyelid cilia d. Hypertelorism. III. Facial bone malformation—the most characteristic findings are as follows a. Hypoplasia of the malar bones i. Often with clefting through the arches. ii. ii. Limited formation of the residual zygomatic complex. b. Orbits i. Hypoplastic lateral aspects of the orbits ii. Dysplastic inferior lateral orbit. c. Maxilla and mandible i. Characteristically hypoplastic ii. Variable effects on the temporomandibular joints iii. Anterior open bite iv. A steep occlusal plane. v. Sleep apnea and sudden infant death syndrome [1, 12, 13].
IV. Nose/mouth
a. Respiratory compromise in severely affected patients as a result of the following two factors: i. Presence of maxillary hypoplasia, which tends to constrict the nasal passages and results in a degree of choanal stenosis or atresia. ii. Presence of mandibular micrognathia and a retro- positioned tongue obstructing the oropharyngeal and hypopharyngeal spaces. b. Nasal deformity c. Microstomia d. Cleft palate with or without cleft lip e. High-arched palate f. Malocclusion g. Open bite.
Differential Diagnosis
a. Nager’s acrofacial dysostosis b. Miller acrofacial dysostosis c. Oculoauriculovertebral spectrum [1]. Diagnostic Tests
- Radiographs and CT for evaluation of craniofacial ab- normality
- Audiological evaluation for hearing impairment
As patient’s main concern was esthetics, the patient was explained regarding the need for pre-surgical orthodontic treatment and right sagittal split osteotomy for mandibular advancement along with genioplasty. Also genetic counseling was provided to the patient.
Review of Literature
Mann and Kilner [14] assumed the etiology to be an inhibitory process occurring towards the seventh week of the embryonic life and affecting the facial bones deriving from the first visceral arch. John Mckenzie suggested that the cause of the abnormality is a defect of the stapedial artery which causes maldevelopment in its own field of as well as in the region of the first visceral arch. Valdez, et al. [15] suggested that haploinsufficiency of treacle in TCS patients might cause insufficient rRNA production in the prefusion neural folds, resulting in abnormal craniofacial development. (The cephalic neural crest cells probably require a higher threshold concentration of rRNA for their survival and proper differentiation during early embryogenesis).
Franceschetti and Klein reviewed the literature and described the typical characteristics of the syndrome as follows [16]: 1) Antimongoloid palpebral fissures with either a notch or coloboma of the outer third of the lower lid, and occasional absence or paucity of the lashes of the lower lid. 2) Facial bone hypoplasia, especially of the malar bones and mandible. 3) Malformation of the external ear, and occasionally of the middle and inner ear, with low implantation of the auricle. 4) Macrostomia, high palate, malocclusion and abnormal position of the teeth. 5) Atypical hair growth in the form of tongue-shaped processes of the hair-line extending towards the cheeks in the pre-auricular region. 6) Association at times with other anomalies, such as obliteration of the naso-frontal angle, pits or clefts between the mouth and ear, and skeletal deformities.
After this description was published, some of these features were regarded as being of lesser importance and some were emphasized in the diagnosis. Thus Axelsson, et al. named the following features as “obligatory” [16]:
- Antimongoloid palpebral fissures.
- Anomaly of the lower lid: coloboma of the outer third, or deficient lashes, or both.
- Hypoplasia of the malar bones.
- Hypoplasia of the mandible.
Mittman [17] described additional features of scarring alopecia and acne keloidalis nuchae in a patient who had some of the classic features of TC S.
Hertle R described ocular findings in 24 patients with TCS. All patients had some eye abnormality like amblyopia, anisometropia, refractive errors, strabismus, lid and adnexal abnormalities and vision loss. [18] Franceschetti and Klein described five clinical forms:
- The complete form (having all known features)
- The incomplete form (presenting variably with less severe ear, eye, zygoma, and mandibular abnormalities)
- The abortive form (only the lower lid seudocoloboma and zygoma hypoplasia are present)
- The unilateral form, (anomalies limited to one side of the face)
- The atypical form (combined with other abnormalities not usually part of the typical syndrome).
In our case, patient presented incomplete form of syndrome.
In a study by Steinbacher DM and Bartlett SP the ramus body angle was elevated to 150.28 degrees as opposed to 114.2 degree in controls [19]. The mandibular plane angle was also significantly elevated in these patients. Elevated antigonial angle and deep notch are indicators of reduced mandibular growth and vertical growth pattern. Even condyles show significant reduction in volume [20]. Zygoma is not fully developed and classified into 3 types: minor form (type 1); moderate (type 2); severe (type 3). Our patient was having type 2 zygoma deformity.
Management
Management of individuals affected by TCS requires a multidisciplinary approach. This involves craniofacial surgeons, orthodontists, ophthalmologists, otolaryngologists and speech pathologists. Depending on the clinical features and severityof manifestations, the patient may require tracheostomy at birth, multiple surgeries to correct eyelid coloboma and cleft palate (in the early years) followed by orbital reconstruction and maxillomandibular osteotomies (at about 5–7 years of age) [21].
The development of speech and language skills depends on the child’s ability to hear during the first 3 years of life. As the great majority of these patients are of normal intelligence, early recognition of deafness and its correction with hearing aids or surgery, when possible, is of great importance for development. An affected parent of either sex will transmit the defect to 50% of his or her offspring in accordance with mendelian laws of genetics. This emphasizes the importance of genetic counseling to affected individuals [17].
Discussion
TCS is a mendalian disorder which favors an autosomal dominant pattern of inheritance; rarely however an autosomal recessive pattern does occur. In a majority of cases it is caused by deletions or insertions in TCOF 1, but a small fraction are associated with mutations in POLR1C and POLR1D genes (recently identified candidate genes).
Both POLR1C and POLR1D are RNA polymerase subunits, which encode RNA polymerase (Pol) I and III subunits that transcribe rRNAs [22, 23]. Mutations of POLR1C and POLR1D causative for TCS are a recent discovery, and there are currently no mammalian models available to study these mutations and their relationship to TCS. However, two groups from America and Hong Kong, have used zebrafish to develop a disease model in the hope of providing insight into the roles of Polr1c and Polr1d [24]. Zebrafish have been shown to be a good model to study facial development, and the formation of facial structure in zebrafish is completed in 5 to 7 days [25, 26]. Zebrafish polr1c and polr1d mutants [27] were used in these studies. Both mutants exhibit the TCS craniofacial malformation, such as the hypoplasia of the ethmoid plate, and jaw agenesis [28]. Alcian blue cartilage staining of the cranial cartilage in 5 days post-fertilization (DPF) polr1c and polr1d homozygote mutants displayed different degrees of hypoplastic phenotypes when compared to their control siblings. In addition, the staining further suggested the parachordal cartilages were not affected in the mutants, which indicated that mesodermally derived tissue had developed normally [24]. On the other hand, development of NCCs was investigated in the mutants via differentin situ hybridization markers. The pre-migratory and migratory NCCs markers, sox10 and foxd3, did not show significant differences in expression [24]; while the early cranial CNCC marker dlx2a, and the neural crest derived ectomesenchyme marker fil1a, showed reduced expression in the pharyngeal arches region [28]. These results indicated the specification and migration of NCCs remains normal in mutants, but there was an overall reduction in the number of NCCs localized in the pharyngeal arches. The mitotic marker phospho-histone H3 (pHH3) staining on NCCs concluded both polr1c and polr1d mutants have a lower cellular proliferation ability. Furthermore, apoptotic cell death in the head region was found in 24 hours postfertilization (hpf) embryos [28]; however, these apoptotic cells are not co-localized with the NCCs in 24 or 48 hpf embryos [24]. To summarize, while the NCCs migration process is normal in both mutants; the higher rate of apoptotic cell death of pre- migratory NCCs, together with decreased proliferation of migrating NCCs results in a reduction in the number of migrating NCCs available for proper pharyngeal arch development and cartilage formation [24].
The typical characteristics of the TCS as stated by Franceschetti and Klein in 1949 are as follows: [22]
- Antimongoloid palpebral fissures with either a notch or coloboma of outer third of the lower lid and occasional absence or paucity of lashes of lower eyelid. Antimongoloid palpebral fissures were observed in the reported case on the right side.
- Hypoplasia of facial bones, especially the malar bones and mandible. These features were present in our case.
- Malformation of the external ear and occasionally middle and inner ear, with low implantation of the auricle. Ear malformation on the right side was observed in our case.
- Macrostomia, high‑arched palate, malocclusion, and abnormal position of the teeth.
- Association at times with other anomalies, such as obliteration of the naso‑frontal angle, pits or clefts between the mouth, and ear and skeletal deformities.
Obliteration of the nasofrontal angle and cleft was not observed in our case.
Many of the clinical features in a patient presenting with TCS were found in our case, however it cannot be termed as a classical case. A multidisciplinary approach is required for the management of individuals affected by TCS involving craniofacial surgeons, orthodontists, ophthalmologists, otolaryngologists, and speech therapists.
Conclusion
In early 2016, a potential new therapy for TCS was reported. A research team combined an in vitro and in vivo model and discovered that Tcof1 haploin sufficiency results in oxidative stress induced neuroepithelial cell death [29]. High levels of endogenous oxidative stress level was found in the neuroepithelium under normal development however, with proper cellular repair systems in place, no excessive cell death was found and normal facial development was achieved. However, in the case of the Tcof1+/− mouse model, the repair machinery was lost and thus resulted in excessive apoptotic cell death and caused facial malformation. A common anti- oxidant, N-acetyl-cysteine (NAC), was applied through intraperitoneal injection of pregnant mothers to suppress the DNA damage and subsequently prevent p53 accumulation and cellular apoptosis. Results showed that such treatment could partially rescue the facial phenotype. However, similar to the p53 inhibition therapy, the Tcof1 mutant pups cannot survive due to other anomalies, such as respiratory failure [29]. Moreover, such outcomes have exposed the limitation of using the zebrafish model that has a totally different respiratory system. However, zebrafish, could provide a unique advantage as a cheap and effective small- molecules screening platform to select potential molecules for further tests in the mammalian system [30].
Some reports have suggested that multiple major reconstructive surgical procedures in severe form of TCS have shown only marginal improvement and these patients are unlikely to benefit from stem cell therapy. Therefore, future research is directed toward preventive aspects and early prenatal detection of this condition.
- This article describes a case reported as 1 in 50,000 live births.
- This case report focuses on the extra‑ and intra‑oral clinical features of TCS, and also elaborates diagnostic aids, both before birth and after birth, which helps in quick diagnosis and thus helps in the further treatment plan.
- As this syndrome has the involvement of many structures, it needs a multidisciplinary approach with the help of which a pediatric dentist can help the patient to get maximum benefit
- In addition to OPG, CT images provide better understanding of facial deformity. Informed Consent: Informed consent was obtained from all individual participants included in the study.
References
-
Chen H (2006) Atlas of genetic diagnosis and counseling. Totowa, New Jersey: Humana Press, pp: 967-971.
-
Dixon MJ, Read AP, Donnai D, Colley A, Dixon J, et al. (1991) The gene for Treacher Collins syndrome maps to the long arm of chromosome 5. Am J Hum Genet 49(1): 17‑22.
-
Chen Y, Guo L, Li CL, Shan J, Xu HS, et al. (2018) Mutation screening of Chinese Treacher Collins syndrome patients identified novel _TCOF1_ mutations. Mol Genet Genomics 293(2): 569-577.
-
(2008) National Organization for Rare Disorders Rare Disease Database.
-
Posnick JC (1997) Treacher Collins syndrome: Perspectives in evaluation and treatment, J Oral Maxillofac Surg 55(10): 1120-1133.
-
Children’s craniofacial association.
-
Kasat V, Baldawa R (2011) Treacher Collins syndrome ‑ A case report and review of literature. J Clin Exp Dent 3(1): e395‑e399.
-
Agrawal SM, Parihar SS, Agrawal MG (2013) Mandibulofacial dysostosis (Treacher Collins syndrome) ‑ A rare case report. J Natl Med Dent Res 1: 34‑37.
-
Dixon MJ (1995) Treacher Collins syndrome. J Med Genet 32(10): 806‑808.
-
Sakai D, Trainor PA (2009) Treacher Collins syndrome: Unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol 41(6): 1229-1232.
-
Trainor PA (2010) Craniofacial birth defects: The role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am J Med Genet A 152A(12): 2984-2994.
-
Marszatek B, Wójcicki P, Kobus K, Trzeciak WH (2002) Clinical features, treatment and genetic background of treacher collins syndrome. J Appl Genet 43(2): 223-233.
-
Martelli-Junior H, Coletta RD, Miranda RT, Barros LM, Swerts MS, et al. (2009) Orofacial features of Treacher Collins syndrome. Med Oral Patol Oral Cir Bucal 14(7): 344-348.
-
Mckenzie J, Craige J (1955) Mandibulofacial dysostosis (Treacher Collins syndrome). Arch Dis Child 30(152): 391-395.
-
Valdez BC, Henning D, So R, Dixon J, Dixon M (2004) The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci USA 101(29): 10709-10714.
-
Farrar JE (1967) Mandibulo-facial dysostosis- a familial study. Br J Ophthalmol 51(2): 132-135.
-
Mittman DL, Rodman OG (1992) Mandibulofacial dysostosis (Treacher Collins syndrome): a case report. J Natl Med Assoc 84(2): 1051-1054.
-
Hertle RW, Ziylan S, Katowitz JA (1993) Ophthalmic features and visual prognosis in the Treacher-Collins syndrome. Br J Ophthalmol 77(10): 642-645.
-
Steinbacher DM, Bartlett SP (2011) Relation of the mandibular body and ramus in Treacher Collins syndrome. J Craniofac Surg 22(1): 302-305.
-
Travieso R, Chang CC, Terner JS, Beckett J, Wong K, et al. (2013) A range of condylar hypoplasia exists in Treacher Collins syndrome. J Oral Maxillofac Surg 71(2): 393-397.
-
Trainor P, Dixon J, Dixon M (2009) Treacher Collins syndrome: etiology, pathogenesis and prevention. Eur J Hum Genet 17(3): 275-283.
-
Alva P, Shetty SR, Babu S (2011) Treacher Collins syndrome ‑ A report of two cases and review of literature. J Clin Exp Dent 3(1): e395‑e399.
-
Laferté A, Favry E, Sentenac A, Riva M, Carles C, et al. (2006) The transcriptional activity of RNA polymerase I is a key determinant for the level of all ribosome components. Genes Dev 20(15): 2030– 2040.
-
Noack Watt KE, Achilleos A, Neben CL, Merrill AE, Trainor PA (2016) The roles of RNA polymerase I and III subunits polr1c and polr1d in craniofacial development and in zebrafish models of treacher collins syndrome. PLoS Genet 12(7): e1006187.
-
Schilling TF, Kimmel CB (1994) Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development 120(3): 483-494.
-
Swartz ME, Sheehan-Rooney K, Dixon MJ, Eberhart JK (2011) Examination of a palatogenic gene program in zebrafish. Dev Dyn 240(9): 2204-2220.
-
Amsterdam A, Burgess S, Golling G, Chen W, Sun Z, et al. (1999) A large-scale insertional mutagenesis screen in zebrafish. Genes Dev 13(20): 2713-2724.
-
Lau MC, Kwong EM, Lai KP, Li JW, Ho JC, et al. (2016) Pathogenesis of POLR1C-dependent Type 3 Treacher Collins syndrome revealed by a zebrafish model. Biochim Biophys Acta 1862(6): 1147-1158.
-
Sakai D, Dixon J, Achilleos A, Dixon M, Trainor PA (2016) Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat Commun 7: 10328.
-
Gut P, Baeza-Raja B, Andersson O, Hasenkamp L, Hsiao J, et al. (2013) Whole-organism screening for gluconeogenesis identifies activators of fasting metabolism. Nat Chem Biol 9(2): 97-104.
- Reconstruction of Nasal Defect Using a Local Flap Based in the Nanoperforants Concept: A Case Report
- Transplant Tourism in Japan: Insights from Nationwide Surveys and Emerging Ethical Challenges
- The Challenge of Peritoneal Vaginoplasty in Transgender Women: Review Literature and Personal Perspectives
- Algorithmic Paternalism: Autonomy Versus Automation
- Dermatoscopy in Gorlin Syndrome: Avoiding the Disfigurement in Patients
- How Artificial Intelligence is Ushering in a New Era of Innovation in General and Plastic Surgery: A Short Communication