Postnatal Diagnosis of Sacral Agenesis with Cerebral Atrophy: A Case Report
Background: Caudal regression syndrome (CRS), also called sacral agenesis is a rare disorder with a spectrum of congenital malformations ranging from simple anal atresia to absence of sacral, lumbar and possibly lower thoracic vertebra. It has an overall incidence of 1 in 60,000 live births. Case Report: We present a postnatal diagnosed case of CRS, delivered at 36 weeks gestation to a multigravida with poorly controlled gestational diabetes. We highlight the associated findings, which include arthrogryposis, cerebebral atrophy, urogenital and cardiac lesions. Conclusion: Sacral agenesis is an uncommon entity; the management is quite challenging, requiring multi-disciplinary intervention. The outcome depends on the spectrum and severity of abnormalities.
Introduction
Caudal regression syndrome (CRS), otherwise known as sacral agenesis is an uncommon disorder with a spectrum of congenital malformations ranging from simple anal atresia to absence of sacral, lumbar and possibly lower thoracic vertebra. It has an overall incidence of 1 in 60,000 live births [1, 2, 3, 4]. It was first described by Geoffroy Saint-Hilaire and Hohl in 1852 [5].
Chromosomal abnormalities, hyperglycemia, vascular hypo perfusion, folic acid deficiency, exposures to trimethoprim-sulfamethoxazole or minoxidil are postulated as contributing factors. Infants of diabetic mothers are known to have a 200-300 fold risk compared to the general newborn population. Other malformations associated with CRS include, urogenital, orthopedic and cardiac. The syndrome commonly arises around the fourth gestational week due to failure in formation of caudal elements of the embryo [2, 3]. Cerebral anomalies are rarely associated with this condition [6].
Diagnosis is usually antenatal, through ultrasound and fetal MRI in the second trimester [4, 7, 8, 9]. Intelligence is generally not affected and the overall outcome depends on the spectrum and severity of malformations present [1, 2, 6].
Case Summary
We present a male neonate, late preterm, born at 36 weeks gestation by spontaneous vaginal delivery, to a multigravida, with poorly controlled gestational diabetes, on insulin. She did not present with any antenatal record and her previous obstetric outcomes were normal. He was vigorous at birth with good APGAR scores and birth weight of 2.7 kg.
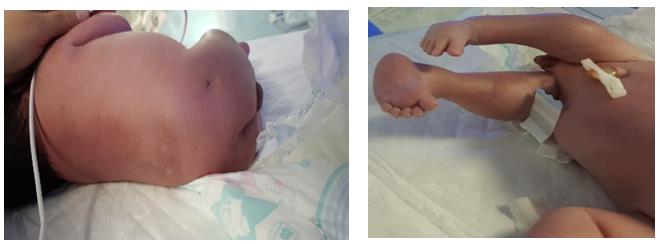
Examination findings at delivery revealed multiple anomalies, particularly affecting the musculoskeletal system. Both lower limbs appeared short and slim with fixed flexion deformity of the hip joints, fixed extended deformed knee joints. No sacral bone was palpable, normal anal opening was seen. He also had multiple dimples over the lower back and buttocks with bilateral fixed talipes equinovarus (Figures 1a & 1b).
Other systemic examination findings include left-sided raised, smooth hemi-scrotum, in keeping with undescended testes. He also had a grade 3 systolic murmur, loudest at left parasternal edge with hepatomegaly.
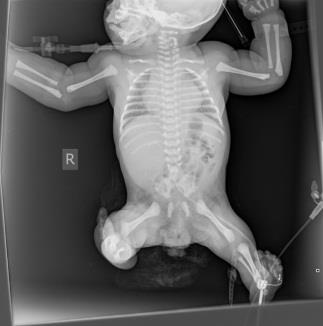
Figure 1(a): Picture of baby showing dimple on the right gluteus and shallow gluteal cleft.
Figure 1(b): Showing fixed flexion at the hips and extension at the knee.
Echocardiography revealed a small sized atrial septal defect (ASD), multiple ventricular septal defects (VSD) and patent ductus arteriosus (PDA). The ultrasound of the abdomen suggested a unilateral polycystic kidney.
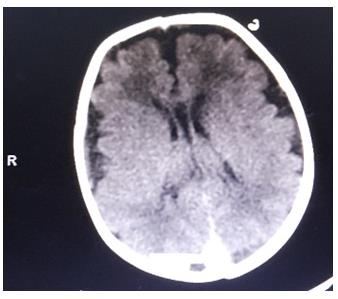
On the second day of life, he was noted to have hypocalcaemia, hypomagnesaemia and clinical seizures. Brain CT showed cerebral atrophy (Figures 2 & 3). He received anticonvulsant therapy and correction of electrolyte derangement with good response. His bowel motion, urination and feeding were essentially normal until discharge.
During his stay, he had multidisciplinary consultation until he was discharged on diuretics and anticonvulsant after 16 days in the unit. His neurologic development so far is within normal limit. The patient remains in follow- up with orthopedic surgery, cardiology, neurology, general surgery and neonatology, in order to keep a close watch on the complications. He is currently awaiting corrective surgery of the lower limb deformities.
Discussion
Caudal regression syndrome is a rare disorder with an incidence of 1: 35,000-60,000 live births. It consists of developmental anomalies of the caudal vertebrae, lower limbs, digestive and urogenital organs. The severity of this syndrome varies from agenesis of the lumbar spine, sacrum, and coccyx to the hypoplasia of the lower extremities. Other anomalies associated with this syndrome are orthopedic, anorectal, vertebral, genitourinary, and cardiac.
Clinically, patients present with a wide spectrum of symptoms ranging from mild motor and sensory deficits in the lower extremities to neurogenic bladder, fecal incontinence and vesicoureteric reflux [1, 2, 3, 4].
Whereas CRS was only detected at birth in our case, prenatal diagnosis is the norm in sacral agenesis with prenatal ultrasound and MRI which demonstrates sacrococcygeal dysgenesis with a high and abrupt termination of the spinal cord with blunt-ending conus medullaris [5, 7, 8, 9, 10]. Prenatal ultrasound also identifies other anomalies, thereby allowing for adequate patient counseling, preparation for delivery and postnatal interventions.
The mother had poorly controlled gestational diabetes in keeping with many documented cases. It is not known if her previous pregnancies with normal outcomes were associated with diabetes mellitus. Persistent maternal hyperglycemia in the first trimester is the most potent teratogen associated with CRS. Sacral agenesis is said to occur in about one percent of diabetic pregnancies [1, 6, 7, 8, 9, 10, 11].
Joint contracture is a well-recognized complication of sacral agenesis, our patient presented with arthrogryposis multiplex congenita (AMC), involving fixed flexion of the hips and fixed extension of the knees. The patient will require extensive orthopedic interventions to achieve ambulation.
Cerebral atrophy and subarachnoid haemorrhage are not normally associated with CRS; neither are they recognized complications of infants of diabetic mother [6, 7]. The immediate post-delivery events in our case did not suggest in-utero hypoxia; however subsequent seizures raise the suspicion of perinatal hypoxia. Whereas patients with sacral agenesis have normal neurodevelopmental outcome, the neurologic outlook for the index patient is guarded. Cerebral Vascular hypo perfusion which is one of the recognized causative factors in CRS may have played a role in the cerebral atrophy observed in this case [2, 3].
Diverse urogenital anomalies are described in patients with sacral agenesis, our case had unilateral polycystic kidney, in addition to unilateral undescended testes. However there was no urinary or faucal incontinence [2-
4]. Our case also had cardiac lesions: small atrial and ventricular septal defect with a patent ductus arteriosus. These lesions have so far been successfully managed medically.
Prenatal diagnosis of CRS is unlikely to have made a difference in our patient as termination of pregnancy is not culturally acceptable in this setting. The index patient will require long term multi-disciplinary management with the achievement of normal ambulation been a great challenge. It is unclear what the neurodevelopmental outcome will be for him.
Conclusion
This case highlights the rarity of CRS as well as the diversity of associated anomalies. The importance of optimal antenatal care cannot be over-emphasized. There is a need to carry out genetic studies in affected families to better understand the interplay between genetic and physical factors in the presentation of this syndrome.
References
-
Semba K (2013) Etiology of Caudal Regression Syndrome. Hum Genet Embryol 3(2): 107.
-
Sen KK, Patel M (2007) Caudal regression syndrome. Med J Armed Forces India 63(2): 178-179.
-
Adra A, Cordero D, Mejides A, Yasin S, Salman F, et al (1994) Caudal regression syndrome: etiopathogenesis, prenatal diagnosis, and perinatal management. Obstet Gynecol Surv 49(7): 508-516.
-
Duncan M, Cantú-SalinasA, Villarreal-Rodríguez D, Muñiz-Landeros C, Villarreal-Velázquez H (2014) Caudal regression syndrome: A case report. Medicina Universitaria 16(63): 45-103.
-
Kumar Y, Gupta N, Hooda K, Sharma P, Sharma S, et al. (2017) Caudal regression syndrome: A case series of a rare congenital anomaly. Pol J Radiol 82: 188-192.
-
Chen CP, Chen CY, Lin CY, Shaw SW, Wang W, et al. (2005) Prenatal diagnosis of concomitant alobar holoprosencephaly and caudal regression syndrome associated with maternal diabetes. Prenat Diagn 25(3): 264-266.
-
Stroustrup Smith A, Grable I, Levine D (2004) Caudal regression syndrome in the fetus of a diabetic mother. Radiology 230(1): 229-233.
-
Aslan H, Yanik H, Celikaslan N, Yildirim G, Ceylan Y (2001) Prenatal diagnosis of caudal regression syndrome: A case report. BMC Pregnancy Childbirth 1(1): 8.
-
Negrete LM, Chung M, Carr SR, Tung GA (2015) In utero diagnosis of caudal regression syndrome. Radiol Case Rep 10(1): 1049.
-
Bouchahda H, El Mhabrech H, Hamouda B, Ghanmi S, Bouchahda R, et al. (2017) Prenatal diagnosis of caudal regression syndrome and omphalocele in a fetus of a diabetic mother. Pan Afr Med J 27: 128.
-
Knight B (2011) Caudal regression syndrome: A case report. AANA Journal 79(4): 281-282.
- Capacity Constraints in Pediatric Inpatient Psychiatric Care: A Cross-Sectional Analysis of Bed Availability and Geographic Access in North Carolina
- Why Healthcare Analytics Still Optimizes the Wrong Things
- Coding, Coverage, and Care: The Infrastructure of Transgender Health Inequities
- The Effect of Classroom Attendance on Academic Achievement of Management and Leadership Discipline of Nursing Students at Instituto Superior Cristal and Universidade de Dili, Timor-Leste, 2024: A Case Study
- The Role of Social Bonds in Facilitating Shared Investments and Resource Allocation: Addressing the “Wrong Pocket Problem” in Public Health and Healthcare
- Social-Cultural Factors Contributing to Antimicrobial Resistance in Livestock Farmers and Community Households in Kayonza District, Rwanda