Inherited Epidermolysis Bullosa: A Clinical Case
Epidermolysis bullosa (EB) is a rare group of disorders, clinically and genetically heterogeneus, characterized by an extreme mucosal fragility that predisposes patients to the development of blisters induced spontaneously or after mimimal contact or friction. These genodermatoses are caused by defects in the proteins involved in the dermoepidermal junction. Currently, more than 1000 mutations and over 20 genes have been identified as responsible for these disorders, with over 30 phenotypically distinct subtypes of EB currently identified. Here, we describe the clinical case of a 37 years old male, diagnosed genetically at age 2 months with inherited epidermolysis bullosa, dystrophic subtype. The results of this study have revealed an alteration in the structural protein type VII collagen, caused by a recessive mutation in the CLO7A1 gene, located in chromosome 3p21.1 In this report we also provide information about the clinical manifestations of each EB major type and subtype, and current recommendations on diagnosis. With the intention of giving visibility to these disorders, we have created a medical documentary entitled “Inherited Epidermolysis Bullosa†that can be seen on you tube at (poner enlace aqui).
Introduction
Epidemolysis bullosa (EB) iS a rare an uncurable disease at the present time, clinically and genetically heterogeneous, characterized by extreme skin and mucosa fragility, with formation of blisters, spontaneously or in response to minor contact/friction.
This genodermatosis is caused by defects in the key proteins (intracellular, extracellular or transmembrane) involved in the dermoepidermic junction. Four main types of EB are distinguished upon morphological analysis of skin samples - simplex, junctional dystrophic and Kindler syndrome – and 30 subtypes are identified based on the clinical severity, inheritance pattern and molecular defect. More than 20 different genes and 1000 mutations account for the genetic and allelic heterogeneity of EB.
Extracutaneous manifestations include anemia, pseudosyndactyly (fusion of fingers and toes), dysphagia (difficulty swallowing), malnutrition, constipation, osteoporosis, muscular dystrophy, renal failure, cancer and more. Therefore, caring for these patients require a multidisciplinary team approach [1].
Our Clinical Case
We describe here the case of a 37 year old man, at the present time, who was first diagnosed with inherited epidermolysis bullosa at 2 months of age. The diagnostic and genetic testing revealed that he has the recessive dystrophic, generalized severe EB subtype. The genetic study also confirmed the presence of an alteration in the structural protein type VII collagen, involved in the cell matrix, caused by a mutation in the COL7A1 gene, located in the chromosome 3p21.1.
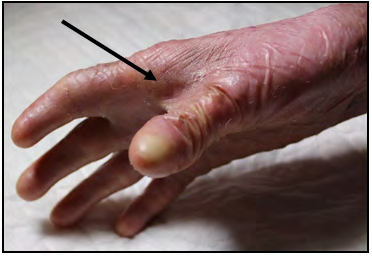
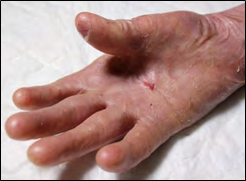
Clinically, the patient presents an extreme fragility in the skin and severe generalized blisters, predominantly located on the skin covering the main joints: elbows, hips, knees and ankles. This chronic tissue disorder has progressively developed into severe contractures and some joint deformities, as well as partial fusions of the interdigital spaces (pseudosyndactyly) in hands and feet (Figures 1-6). At the present time, the patient is able to personally perform all the treatment for the skin lesions, placing dressings on the arms and legs every 48 hours, with materials not covered by the public health insurance. After an exhaustive cleaning of each lesion with gauzes and saline solution, he places a topical healing cream and the dressings.
The extracutaneous manifestations in our patient involve recurrent episodes of stenosis of the esophagus, which requires periodic esophagic dilations with an air-filled balloon that is passed through an endoscope. He also presents chronic renal failure, which irreversibly has required a kidney transplant and he describe having episodes of paroxysmal dyspnea (shortness of breath) due to the severe affectation of the mucosa. Additionally, this patient developed anemia, malnutrition and growth retardation during his infancy and adolescence.
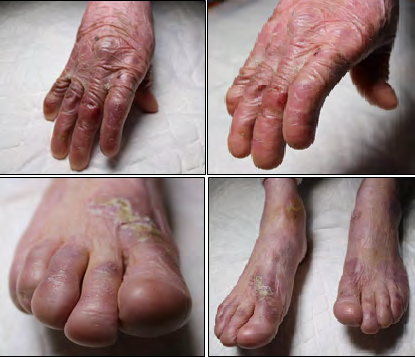
Figures 1-4:Atrophic cutaneous scars and loss of nails in fingers and toes.
Discussion
Epidermolysis bullosa (EB), commonly called “butterfly skin”, consist of a group of clinically and genetically heterogeneous rare disorders, characterized by an extreme cutaneous and mucosal fragility, which highly predisposes patients highly to the development of blisters, spontaneously or after minimal friction. To date, management of this disease is only systematic and it is considered devastating and incurable. EB affects individuals of all ethnicities and it is not sex-linked. Currently and according to DEBRA (Dystrophic Epidermolysis Bullosa Research Association) its incidence is 15-19 cases per 1,000,000 live births, with a prevalence of 10 per million. In Spain there are approximately 500 affected individuals.
Immunological and genetic studies have determined that EB is caused by autosomal mutations, either dominant or recessive, in the genes that codify for structural proteins involved in the integrity and mechanical estability of the skin. These proteins can be intracellular, transmembrane or extracellular, and be involved in the formation of the cytoskeleton, cell-to-cell anchorage or intercellular matrix. Due to the great number of proteins involved in the cleavage of our epidermis and its genetic pattern, this disease presents a high clinical heterogeneity, with over 30 phenotypically and genetically distinct nosological entities described, more than 1000 possible mutations and at least 20 genes involved.
Inherited Epidermolysis Bullosa is also characterized by extracutaneous manifestations that include: nail and hair anomalies (alopecia), intrabuccal blisters, dental complications (tooth loss, microstomia, ankyloglossia) and eye and skin infections. Patients usually show greater susceptibility to sepsis due to decreased resistance to infections, which along with malnutrition and anemia, increases the morbidity and mortality of these type of disorders.
Major EB Types and Subtypes
At the present time and based on decades of clinical analyses, four major types of inherited epidermolysis bullosa are recognized, considering the site of rupture within the epidermis: EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler Syndrome. In EBS, the blisters form at the intraepidermic level, within the middle/upper epidermal layers (which could affect the suprabasal or the basal keratinocytes). In JEB theseparation develops within the lamina lucida and in DEB they occur within the sublamina densa region within the uppermost dermis.
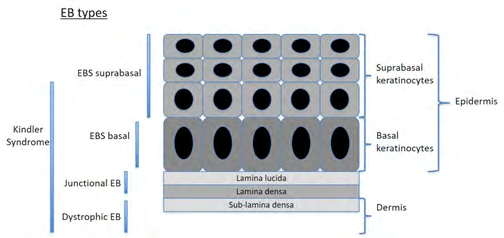
In 2013, Fine, et al. [2] created an international consensus report to facilitate the diagnosis and classification of EB, based on newer data, both clinical and molecular. A new approach named “onion skinning”, because of its analogy to peeling an onion, is proposed for the classification of patients with EB. Schematically, this progressive approach may be summarized as taking into consideration first, the major type of EB based on site of cleavage (described in Figure 7, followed by the identification of the phenotype of each type and subtype (severity and distribution), mode of transmission, ultrastructural site of cleavage, protein involved, gene involved and mutation type. Following these criteria, over 30 subtypes of EB have been described.
Epidermoysis Bullosa Simplex (EBS)
EBS is the most common type of EB, representing a 75-85% of all the cases in Western countries [3]. It is characterized by the formation of blisters in the skin, mostly induced by light friction, with localized or generalized anatomical distribution and that usually disappear without scarring. Its inheritance is autosomal dominant in 95% of the cases, with also some documented rare cases of autosomal recessive inheritance [3, 4, 5, 6, 7].
Mostly, EBS is caused by mutations in genes involved in the production of keratin, resulting in the formation of a cleavage at the level of the basal keratinocytes, originating blisters at the intraepidermic level. The rare EB variants are asociated with mutations in genes that codify for other structural proteins, such as desmoplakin, plakophilin-1, plakoglobin, integrin α6β4, collagen type XVII, plectin, transglutaminase-5 and dystonin [8].
EBS can be subdivided in two types, basal and suprabasal, considering wether the cutaneous fragility is originated within the basal keratinocytes or in the middle/upper layers of the epidermis, respectively.
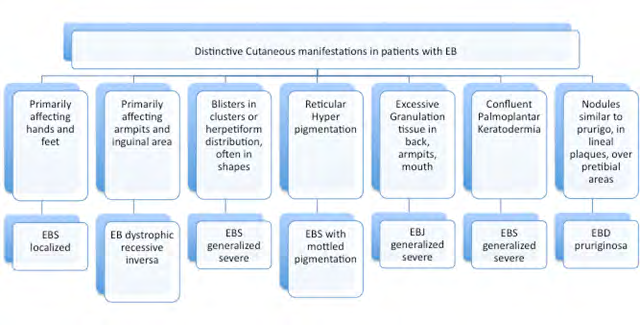
The most common clinical subtypes at the basal level are: localized EBS, generalized EBS and generalized intermediate (Table 1). Localized EBS is the most mild form, generally developing in between infancy and the third decade of age, characterized by blisters on the palms of the hands and the bottoms of the feet [9]. Ulceration of the mucosa, provoked by microtraumatisms due to sucking on baby bottles, are generally resolved with age, without scarring of Milia’s cysts. Hair and teeth are normal, and ungueal dystrophy is mild and rare. Generalized EBS is the most severe form of EBS. It appears soon after birth, with disseminated topography and it is associated with high morbimortality during the perinatal and infancy periods. Its more distinguishing feature is the formation of blisters in clusters, in an archeous (herpetiform) distribution. These blisters can appear spontaneously in the trunk, upper extremities or neck. The oral mucosa is also frequently affected. Palmoplantar hyperkeratosis (thickening) appears also during infancy and can progress overtime to confluent keratoderma. Additional clinical manifestations include ungual dystrophy, loss of nails and hair (telogen effluvium), atrophic scarring, Milia’s cysts, laryngeal stenosis, anemia and growth retardation. This subtype of EBS tends to become mild with age, although specfic environmental conditions, such as high temperatures or sweating during summer, can act as triggering factors [10]. Generalized intermediate EBS is characterized by the formation of non-herpetiform blisters soon after birth or during the first infancy, usually mild and affecting mostly hands, feet and extremities. The development of the hair, teeth and nails are normal, and anemia and growth retardation are rare [11].
The rare types of Basal EBS include: with mottled pigmentation (EBS-MP), migratory circinate (EBS-Migr), autosomal recessive K14 (EBS-AR K14), With pyloric atresia (EBS-PA), With muscular dystrophy (EBS-MD), EBS of Ogna (EBS-Og), ExophIlin-5 Deficiency (EBS-AR exophilin 5), BP230 Deficiency (EBS-AR BP230). Recently, a new protein, Kelch-like 24 (KLHL24) has been identified, and its alteration induce excessive ubiquitination and fragmentation of keratin 14. This mutation is associated with a subtype of basal EBS with distinct clinical manifestations (alopecia, follicular and cutaneous atrophy) [12].
At the suprabasal level, several subtypes of EBS are recognized: Acral peeling skin syndrome (APSS), EBS superficialis, acantholytic (EBS-acanth) and skin fragility syndromes due to deficiencies in the structural proteins desmoplakin (fragility-woolly hair syndrome), plakoglobin (fragility-plakoglobin deficiency) and plakophilin 1 (fragility- ectodermal dysplasia syndrome) [1].
| Clinical Subtypes (Heredity) | Protein | Mutated Gene | Chromosome | |
|---|---|---|---|---|
| Suprabasal EBS | Acral peeling skin syndrome (ARH) | Transglutaminase 5 | TGM5 | 15q15.2 |
| Suprabasal EBS | Superficialis(EBSS) (unknown) | Unknown | ||
| Suprabasal EBS | Acantholytic (ARH) | Desmoplakin | DSP | 6p24,17q21 |
| Suprabasal EBS | Acantholytic (ARH) | Plakoglobin | JUP | 6p24,17q21 |
| Suprabasal EBS | Skin fragility syndromes: | |||
| Suprabasal EBS | - EBS-desmoplakin (ARH) | Desmoplakin | DSP | 6p24 |
| Suprabasal EBS | - EBS-plakoglobin (ARH) | Plakoglobin | JUP | 17q21 |
| Suprabasal EBS | - EBS-plakophilin (ARH) | Plakophilin 1 | PKP1 | 1q32 |
| Basal EBS | Localized (ADH) | |||
| Basal EBS | Generalized severe (ADH) | Keratin 5 | KRT5 | 12q13.13 |
| Basal EBS | Generalized intermediate (ADH) | Keratin 14 | KRT14 | 17q21.2 |
| Basal EBS | With mottled pigmentation (ADH) | Keratin 5 | KRT5 | 12q13.13 |
| Basal EBS | Migratory circinate | Keratin 5 | KRT5 | 12q13.13 |
| Basal EBS | Autosomal recessive K14 (ARH) | Keratin 14 | KRT14 | 17q21.2 |
| Basal EBS | With muscular dystrophy (ARH) | Plectin | PLEC | 8q24 |
| Basal EBS | With pyloric atresia (ARH) | Plectin | PLEC | 8q24 |
| Basal EBS | With pyloric atresia (ARH) | α6β4 Integrin | ITGA6 | 2q31.3 |
| Basal EBS | With pyloric atresia (ARH) | ITGB4 | 17q25 | |
| Basal EBS | Ogna (ADH) | Plectin | PLEC | 8q24 |
| Basal EBS | Autosomal recessive-BP230 deficiency (ARH) | Bullouspemphigoid antigen-1 (BP230) | DST | 6p12.1 |
| Basal EBS | Autosomal recessive-exophilin 5 deficiency (ARH) | Exophilin 5 | EXPH5 | 11q22.3 |
| Basal EBS | Due to KLHL24 (ADH) | Protein 24 similar to kelch | KLHL24 | 3q27.1 |
Table 1: Clinical subtypes of Epidermolysis Bullosa Simplex (EBS), heredity, proteins involved, mutated genes and chromosome loca
Junctional Epidermolysis Bullosa (JEB)
JEB is, in the majority of cases, caused by mutations in the LAMB3 gene, which encodes laminin-5, a protein involved in formation of the anchoring filaments located in the junction between the lamina lucida and the sub-lamina densa, producing the characteristic intra-lamina cleavage of blisters. This disorder, of autosomal recessive heredity, is also characterized by lesions in the skin and mucosa, with scar formation after healing. A distinctive characteristic is the appearance of enamel hypoplasia and the development of cavities, due to mutations in genes involved in dental histomorphogenesis [13]. The rare variants of JEB are associated with mutations in genes that codify for the hemidesmosomal proteins collagen XVII, integrin α6β4 and integrin α3.
JEB is subdivided in two major subtypes: generalized JEB and localized JEB (Table 2). Generalized Severe JEB appears from birth and can affect many regions of the skin. A pathognomonic sign is the appearance of granulation tissue, with soft, pink bumpy skin erosions, often seen around the lips, face, eyes, nose, superior region of the back,armpits and inguinal folds [14]. The clinical manifestations include intraoral blisters, microstomia (small mouth) and ankyloglossia (tongue-tie), but not as severe as in the recessive forms of EBD. Other commonly found features are the appearance of thick, yellow and curved nails, with longitudinal striations (onychogryphosis) or the absence of nails (anonychia) due to atrophy and cicatrization of the ungual bed and matrix.
At the extracutaneous level, erosions and blisters can appear in all the squamous stratified epithelial tissues, including the conjunctival, oral, corneal, gastrointestinal, tracheobronchial, pharyngeal, esophageal, rectal and genitourinary mucosa. As a consequence of this, hoarseness and inspiratory stridor are frequent, as well as urethral meatal stenosis and urinary retention. Patients with this type of JEB present growth retardation and multifactorial anemia, and are at high risk of death from pneumonia, infections, sepsis and airway obstructions [1]. Childhood mortality from this condition is the highest until the age of 2. Generalized Intermediate JEB is the most common subtype of junctural EB. Present from birth, involves the appearance of blisters, atrophic scarring, dystrophic or absent nails. Patients usually present postinflammatory hypopigmentation or depigmentation, with cicatricial alopecia (scarring hair loss) in some cases [1].
This subtype of JEB is also associated to growth retardation and anemia [15], causing a high risk for childhood mortality, although less than the Severe subtype. Patients who survive infancy show clinical improvement with age, but are at a higher risk to develop squamous cell carcinoma [16, 17]. A third type of generalized JEB, with pyloric atresia, it is characterized for, in addition to producing generalized blisters from birth, the appearance of congenic pyloric occlusion, causing anomalies in the genitourinary tract with a highly variable prognosis (1). Localized JEB
are mostly characterized by mild to severe blistering in the palms of hands, sole of feet, elbows and knees. Additionally, the Inversa JEB subtype involves the development of severe blisters confined to intertriginous skin sites, the esophagus and vagina (1). A third subtype of JEB, named LOC Syndrome, is associated with an altered cry in the neonatal period, the formation of blisters and scars in the face and neck and the production of granulation tissue affecting in particular the upper airway tract (larynx), conjunctiva, nails and periungual/subungual sites [1].
| Clinical Subtypes (Heredity) | Protein | Mutated Gene | Chromosome | ||
|---|---|---|---|---|---|
| JEB Generalized | Generalized severe (ARH) | Laminin-332 | LAMA3 | 18q11.2 | |
| JEB Generalized | Generalized severe (ARH) | Laminin-332 | LAMB3 | 1q32.2 | |
| JEB Generalized | Generalized intermediate (ARH) | Laminin-332 | LAMA3 | 18q11.2 | |
| JEB Generalized | Generalized intermediate (ARH) | Laminin-332 | LAMB3 | 1q32.2 | |
| JEB Generalized | With pyloric atresia (ARH) | Integrin α6β4 | LAMC2 | 1q25-q31 | |
| JEB Generalized | With pyloric atresia (ARH) | Integrin α6β4 | COL17A1 | 10q24.3 | |
| Late onset | Collagen XVII | COL17A1 | |||
| With respiratory and renal involvement | Integrin α3 subunit | ITGA3 | |||
| JEB Localized | Localized (ARH) | Laminin-332 | LAMA3 | 18q11.2 | |
| JEB Localized | Localized (ARH) | Laminin-332 | LAMB3 | 1q32.2 | |
| JEB Localized | Localized (ARH) | Laminin-332 | LAMC2 | 1q25-q31 | |
| JEB Localized | Localized (ARH) | Laminin-332 | COL17A1 ITGA6 | 10q24.3 | |
| JEB Localized | Collagen XVII | ITGB4 | 2q31.3 | ||
| JEB Localized | Collagen XVII | ITGB4 | 2q31.3 | Integrinα6β4 | 17q25 |
| Inversa (ARH) | Laminin-332 | LAMA3 LAMB3 | 18q11.2 | Integrinα6β4 | 17q25 |
| Inversa (ARH) | Laminin-332 | LAMC2 | 1q32.2 | ||
| LOC Syndrome (ARH) | Laminin-332, isoform α3 chain | LAM3A |
Table 2: Clinical subtypes of Junctional Epidermolysis Bullosa (JEB), heredity, proteins involved, mutated genes and chromosome l
Dystrophic Epidermolysis Bullosa (DEB)
DEB is characterized by skin fragility and blisters that heal with scrs, changes in nails and formation of millium due to damage to hair follicles. DEB is subdivided in two subtypes, DEB dominant and DEB recessive, based on its inheritance. It is caused by mutations in the COL7A1 gene (localized in chromosome 3p21.31) that codifies for the alpha-1 chain of type VII collagen. This produces a breakage in the tissue under the lamina densa in the basal membrane, forming blisters in the dermic sub-basal lamina. Type VII collagen is the principal component of the anchoring fibrils in this region, and its defective structure is associated with an extreme skin fragility, extensive scarring , flexural contractures of the extremities, joint deformities, mucosa severely affected , malnutrition and growth retardation. Generalized dominant DEB is the is the most severe in this subgroup. It is characterized by the presence of blisters from birth or during childhood, distributed across the whole body, asociated with time to Millia,atrophy, scars and dystrophic nails. It is frequent the appearance of recurrent esophageal blisters and lesions [1].
Generalized recessive DEB is the subtype present in our patient. It represents the most severe and common subtype of recessive EBS. It usually shows clinical manifestations from birth or a little later, with the appearance of blisters that extend progressively to the whole body and heal with atrophic scars [1]. Pseudosyndactyly (partial fusion of interdigital spaces) due to continuous formation of blisters and scars in hands and feet, is a characteristic sign, which can lead towards a complete fusion of all the fingers, forming what is called “boxing glove hands”. Additionally, patients develop excessivecavities, severe ankyloglossia and microstomia, associated to a deficit in the ingestion of nutrients and malnutrition. Other extracutaneous manifestations are corneal blisters, esophageal stenosis, cronic renal failure, multifactorial anemia and growth retardation [1]. Affected individuals who survived childhood have also a significant risk of developing metastatic squamous cell carcinoma, mostly in the areas of persistent chronic lesions, causing premature death due to the specific associated mutagenesis [18]. Although uncommon, a small percentage of these patients can develop dilated cardiomyopathy and congestive heart failure, possibly fatal [19]. Generalized recessive Intermediate DEB is characterized by the formation of less severe blisters, and in general does not cause growth retardation nor anemia, with less frequency of esophageal stenosis and skin affectation, but the risk of carcinoma in the squamous cells is still high [20].
The less frequent subtypes of DEB include variants such as pretibial DEB, which appears in adolescence or in adults, with the formation of blisters, lesions, milia and atrophic scars, that often become violaceus papules, lichenoid and pruritous [21], pruriginosa DEB, which produces skin blisters and uncurable pruritus that worsen with the development of papules, nodules, lichenoid and hypertrophic lesions [22] and DEB of the newborn, where the generalized blistering appear with focal scarring at birth and regresses within the first 6 to 24 months of life.
| Clinical Subtypes | Protein | Mutated Gene | Chromosome | |
|---|---|---|---|---|
| EBD Dominante (ADH) | Generalized | Collagen VII | COL7A1 | 3p21.1 |
| EBD Dominante (ADH) | Localized | Collagen VII | COL7A1 | 3p21.1 |
| EBD Dominante (ADH) | Acral | Collagen VII | COL7A1 | 3p21.1 |
| EBD Dominante (ADH) | Pretibial | Collagen VII | COL7A1 | 3p21.1 |
| EBD Dominante (ADH) | Pruriginosa | Collagen VII | COL7A1 | 3p21.1 |
| EBD Dominante (ADH) | Nails only | Collagen VII | COL7A1 | 3p21.1 |
| EBD Dominante (ADH) | Of the Newborn | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Generalized severe | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Generalized intermediate | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Localized | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Pretibial | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Pruriginosa | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Inverse | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Centripetal | Collagen VII | COL7A1 | 3p21.1 |
| EBD Recesiva (ARH) | Of the Newborn | |||
Table 3: Clinical subtypes of Dyatrophyc Epidermolysis Bullosa (DBS), heredity, proteins involved, mutated genes and chromosome l
Kindler Syndrome (KS)
Of autosomal recessive heredity, KS is caused by mutations in the FERMT1 (KIND1) gene, located in chromosome 20p12.3, which codifies for the homologous protein Kindlin-1, in the fermitin family (FFH1). This can result in ruptures at multiple cleavage planes (intradermal, junctional or sub-lamina densa). Kindlin-1 protein is involved in focal adhesion and it is predominantly expressed in the basal keratinocytes of the skin, periodontal tissues and colon. Therefore its long term deficiency is associated with an increase in the risk of mucocutaneous cancer [23, 24, 25].
KS is a rare dermatosis that is present at birth as blisters generalized distributed, but mainly acrally and that often disappear over the years [26]. In late childhood, SK is characterized by the appearance of a progressive development of poikiloderma (a combination of cutaneous atrophy, depigmentation and telangiectasia), photosensitivity, keratoderma and early skin aging. Tissues of the mucosa are also affected with age, with gingivitis and periodontitis as the most common manifestations. Other complications involve stenosis of the esophagus or genitourinary, gastrointestinal complications, such as colitis or constipation, or the development of skin cancer, carcinoma or melanoma, as adults [27] (Figure 8).
Recommended Diagnostic Testing in EB
When cutaneous fragility is present in a patient (recurrent skin erosions and blisters that appear easily after light injuries), the physician should suspect the existence of an alteration in the proteins involved in the dermoepidermic cleavage, and it is recommended to follow three key steps for the appropriate diagnosis [1].
Clinical Diagnosis
The first step consists in the evaluation of the clinical manifestations in a patient, creating a clinical history that takes into account the distribution and severity of the cutaneous and extracutaneous lesions, date of appearance of symptoms, and the immediate family history (parents’ age, possible consanguinity, geographic origin of the family). In newborns, the clinical diagnosis of the subtype of EB is complex and difficult due to the fact that many of the initial symptoms are common to all four main subtypes. However in the adult patient, the symptomatology of this disorder is generally well established and the clinical manifestations tend to be very specific, allowing to obtain a more precise identification of the specific subtype.
It is also fundamental to perform a differential diagnosis to distinguish between the inherited and the non-inherited EB types. Non-genetic epidermolysis bullose is a rare autoimmune disorder caused by the production by the body of antibodies against specific molecules in the dermoepidermic junction (anchoring fibrils), causing lesions and inflammation. In this case, however, there is not formation of altered proteins like in the genetic types of EB.
Diagnostic Testing
Involves the identification of the level of skin cleavage via antigen mapping with immunofluorescence (IFM) as well as the genetic testing and diagnosis. A skin sample is harvested (biopsy) from an area with fresh (spontaneous or traction-induced) blisters, and the level of skin cleavage is identified via immunofluorescent antigen mapping and/or transmission electron microscopy, facilitating the diagnosis towards one of the four main types of EB, delimitating also the identification of the candidategene. For the genetic testing, a blood sample is used to extract the genomic DNA and detect the specific genetic mutation. The antigen mapping involves the use of of a cluster of antibodies directed against type IV collagen (protein present in the lamina densa at the dermoepidermic junction) in addition to the use of additional specific antibodies against other proteins involved in EB. The antibody against type IV collagen allows for the unequivocal identification of DEB (antigen present at the superior region of the blister) and KS (reduplication of basal membrane).
Thi santibody does not distinguish however, between SEB and JEB, since in both cases the antigen is located at the inferior region of the blister. The use of other specific antibodies against other proteins allow us to differentiate SEB, JEB and DEB, in order to determine the candidate gene. In some cases, the results from the immunofluorescence mapping permit a most precise subclassification, due to the fact that a variation in the intensity of antigen staining (or its total absence) of specific structural proteins, indicates the presence of a mutation within its associated gene. Currently, despite the increase in genetic analysis, IFM is still recommended as a prior step to help identify the level of cleavage and the affected structural protein.
The genetic analysis is initiated with a search for the most common mutations and finalized with the complete sequence of the gene, if necessary. Over the past years, genetic sequencing has advanced in giant steps, from first generation sequencers (Sanger Method) to Next-Generation Sequencing or NGS, which allows multigenic sequencing [29]. These second generation sequencers that appeared around the year 2005, can perform multiple clonal and random copies of DNA fragments of 200-300 bp followed by sequencing similar to what the classic methods used to do to analyze only one or two genes [30]. This advance is very useful for EB as it allows for the design of a direct molecular study specific for the individual, independently from the antigen mapping. In 2018, in a study done by Has, et al. [31]. 40 cases of suspected EB were analyzed by antigen mapping (IFM) and/ or targeted next-generation sequencing (NGS). Their results indicated that IFM detected correctly 76% of the cases, whereas the molecular fingerprinting provided the most accurate detection, in 90% of the cases, finding mutations in genes not previously recognized. This estudy emphasized the efficacy of combining NGS with IFM to resolve unusual EB phenotypes. Additionally, NGS in unable to identify all cases of pathogenic mutations (deletions, duplications) and other complementary techniques such as multiplex ligation probe amplification (mMLPA) and comparative genomic hybridization (array-CGH) are also being used.
It is important to clarify that the mutation analysis of the specific EB is important to provide the best genetic counseling by the clinician, as it provides information about the mode of inheritance, as well as allows for the elaboration of a theoretical and accurate clinical prognostication for the progress of the disease overtime. However, reliance solely on mutational findings must be done with care, because as we know, environmental and modifying genetic factors (epigenetics) can influence the expression of the mutation and create considerable variation in disease severity [2].
Genetic Counseling
This step involves genetic counseling, once the responsible mutation is identified, to establish an heritability profile for the patient and his/her family, to calculate the probability of risk and recurrence in future children and siblings. At this point, a prenatal diagnosis (invasive and non- invasive) could be performed as well as a preimplantational genetic diagnosis [1]. The analysis of mutations on fetal DNA has high validity, representing the best current method for prenatal diagnosis, having replaced the technique of biopsy of fetal skin [7]. This initial exam determines the pattern of heritability and establishes a confidence value in the prenatal testing [6]. The fetal DNA is obtained from the chorionic villi (during the first trimester of pregnancy) or from amniotic cells (after 15 weeks) and is analyzed to detect the possible mutations.
The preimplantation genetic diagnosis (PGD) is a highly specialized procedure, available only in a limited number of centers in the world. This test includes the genetic analysis of one embryonic cell after in vitro fertilization, at the stage of 6-10 cells (3 days). The mutation-free embryos can then be implanted in the uterus by 4-5 day of development [32].
Final Considerations
Although hereditary EB is a rare dermatological disorder, its impact on the lives of the patients and their families is immense, bringing many limitations to the individualas well as physical and emotional suffering. The current absence of effective therapeutic measures, makes essential the need of a successful monitoring of patients with EB, which should imply the contribution of a multi-professional clinical team, generally consisting of a dermatologist, a nurse specialized in this disorder, a primary care physician, a physical therapist, a nutritionist, a psychologist and a social worker. The most appropriate approach to do this is through a specialized network that includes information and specialized centers, and daily caregivers to provide physical and psychological comfort to the patient.
Currently there is not cure for epidermolysis bullosa and local measures (appropriate cleaning of the lesions) and prophylaxis of clinical complications are the only effective strategies to control the symptoms of this disease [33]. However, new therapeutic strategies are currently being evaluated, and hope for these patients depends on ongoing studies, such as the substitution of the damaged structural proteins (recombinant protein infusion) or the mutated genes (gene therapy) as well as allogeneic transplantation of bone marrow, pluripotent stem cells or umbilical cord cells [34].
References
-
Sánchez-Jimeno C, Escámez MJ, Ayuso C, Trujillo-Tiebas MJ, Del Río M en representación de la Cátedra de la Fundación Jiménez Díaz de Medicina Regenerativa y Bioingeniería Tisular DEBR-Eydops (2018) Genetic diagnosis of epidermolysis bullosa: recommendations from an expert Spanish research group. Actas Dermosifiliogr 109(2): 104-122.
-
Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, et al. (2014) Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 70(6): 1103-1126.
-
Abu Sa’d J, Indelman M, Pfendner E, Falik-Zaccai TC, Mizrachi-Koren M, et al. (2006) Molecular epidemiology of hereditary epidermolysis bullosa in a Middle Eastern population. J Invest Dermatol 126(4): 777-781.
-
Ciubotaru D, Bergman R, Baty D, Indelman M, Pfendner E, et al. (2003) Epidermolysis bullosa simplex in Israel: clinical and genetic features. Arch Dermatol 139(4): 498-505.
-
Groves RW, Liu L, Dopping-Hepenstal PJ, Markus HS, Lovell PA, et al. (2010) A homozygous nonsense mutation within the dystonin gene coding for the coiled- coil domain of the epithelial isoform of BPAG1 underlies a new subtype of autosomal recessive epidermolysis bullosa simplex. J Invest Dermatol 130(6): 1551-1557.
-
García M, Santiago JL, Terrón A, Hernández-Martín A, Vicente A, et al. (2011) Two novel recessive mutations in KRT14 identified in a cohort of 21 Spanish families with epidermolysis bullosa simplex. Br J Dermatol 165(3): 683-692.
-
Pigors M, Kiritsi D, Krümpelmann S, Wagner N, He Y, et al. (2011) Lack of plakoglobin leads to lethal congenital epidermolysis bullosa: a novel clinico-genetic entity. Hum Mol Genet 20(9): 1811-1819.
-
Has C, Spartà G, Kiritsi D, Weibel L, Moeller A, et al. (2012) Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med 366(16): 1508-1514.
-
Sprecher E (2010) Epidermolysis bullosa simplex. Dermatol Clin 28(1):23-32.
-
Prodinger C, Diem A, Bauer JW, Laimer M (2016) Mucosal manifestations of epidermolysis bullosa: Clinical presentation and management. Hautarzt 67(10): 806- 815.
-
Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner- Tuderman L, et al. (2008) The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 58(6): 931-950.
-
Lin Z, Li S, Feng C, Yang S, Wang H, et al. (2016) Stabilizing mutations of KLHL24 ubiquitin ligase causes loss of keratin 14 and human skin fragility. Nature Genetics 48(12): 1508-1516.
-
Fine JD, Mellerio JE (2009) Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol 61(3): 367-384.
-
Laimer M, Lanschuetzer CM, Diem A, Bauer JW (2010) Herlitz junctional epidermolysis bullosa. Dermatol Clin 28(1): 55-60.
-
Fine JD, Johnson LB, Weiner M, Suchindran C (2008) Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr 152(2): 276-280.
-
Mallipeddi R, Keane FM, McGrath JA, Mayou BJ, Eady RA (2004) Increased risk of squamous cell carcinoma in junctional epidermolysis bullosa. J Eur Acad Dermatol Venereol 18(5): 521-526.
-
Yuen WY, Jonkman MF (2011) Risk of squamous cell carcinoma in junctional epidermolysis bullosa, non- Herlitz type: report of 7 cases and a review of the literature. J Am Acad Dermatol 65(4): 780-789.
-
Cho RJ, Alexandrov LB, den Breems NY, Atanasova VS, Farshchian M, et al. (2018) APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci Transl Med 10(455).
-
Fine JD, Hall M, Weiner M, Li KP, Suchindran C (2008) The risk of cardiomyopathy in inherited epidermolysis bullosa. Br J Dermatol 159(3): 677-682.
-
Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C (2009) Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986- 2006. J Am Acad Dermatol 60(2): 203-211.
-
Rizzo C, Anandasabapathy N, Walters RF, Rosenman K, Kamino H, et al. (2008) Pretibial epidermolysis bullosa. Dermatol Online J 14(10): 26.
-
Brick K, Hand JL, Frankel AS, Siegel DH, Thomas KB, et al. (2012) Epidermolysis bullosa pruriginosa: further clarification of the phenotype. Pediatr Dermatol 29(6): 732-737.
-
Lahn M, Kloeker S, Berry BS (2005) TGF-beta inhibitors for the treatment of cancer. Expert Opin Investig Drugs 14(6): 629-643.
-
Yasukawa K, Sato-Matsumura KC, McMillan J, Tsuchiya K, Shimizu H (2002) Exclusion of COL7A1 mutation in Kindler syndrome. J Am Acad Dermatol 46(3): 447-450.
-
Kloeker S, Major MB, Calderwood DA, Ginsberg MH, Jones DA, et al. (2004) The Kindler syndrome protein is regulated by transforming growth factor-beta and involved in integrin-mediated adhesion. J Biol Chem 279(8): 6824-6833.
-
Lai-Cheong JE, McGrath JA (2010) Kindler syndrome. Dermatol Clin 28(1): 119-124.
-
Has C, Castiglia D, del Rio M, Diez MG, Piccinni E, et al. (2011) Kindler syndrome: extension of FERMT1 mutational spectrum and natural history. Hum Mutat 32(11): 1204-1212.
-
Jean L Bolognia, Julie V Schaffer, Cerroni L (2018) Dermatología 4th (Edn.), Elsevier Health Sciences, pp: 2880.
-
Has C, Fischer J (2019) Inherited epidermolysis bullosa: New diagnostics and new clinical phenotypes. Exp Dermatol 28(10): 1146-1152.
-
Guan YF, Li GR, Wang RJ, Yi YT, Yang L, et al. (2012) Application of next-generation sequencing in clinical oncology to advance personalized treatment of cancer. Chin J Cancer 31(10): 463-470.
-
Has C, Küsel J, Reimer A, Hoffmann J, Schauer F, et al. (2018) The Position of Targeted Next-generation Sequencing in Epidermolysis Bullosa Diagnosis. Acta Derm Venereol 98(4): 437-440.
-
Lane EB, McLean WH (2004) Keratins and skin disorders. J Pathol 204(4): 355-366.
-
Boeira VL, Souza ES, Rocha Bde O, Oliveira PD, Oliveira MF, et al. (2013) Inherited epidermolysis bullosa: clinical and therapeutic aspects. An Bras Dermatol 88(2): 185- 198.
-
Chiaverini C, Bourrat E, Mazereeuw-Hautier J, Hadj-Rabia S, Bodemer C, et al. (2017) Hereditary epidermolysis bullosa: French national guidelines (PNDS) for diagnosis and treatment. Ann Dermatol Venereol 144(1): 6-35.
- Psychogenic Erectile Dysfunction in Late Adulthood: A Case Report on Clinical Intervention and Intimacy Restoration
- Clinical Trials on COVID-19 in 2025: A New Chapter in Global Health Research
- Innovations and Challenges in Contemporary Medical Clinical Trials: An Editorial Perspective
- Innovations and Challenges in Contemporary Medical Clinical Trials: A Critical Perspective
- Reimagining Clinical Trials: The Power of Continuous Feedback from Medical Reports
- Factors Influencing Brain Drain: Perspectives from a Medical School in Turkey