Curing Systemic Lupus Erythematosus and Severe Aplastic Anemia with an Allogeneic Transplant A Case Report and Brief Literature Review
Systemic lupus erythematosus (SLE) is an autoimmune disease that can affect many organs, including the bone marrow. Aplastic anemia (AA) is a rare but serious complication of this condition. Patients with AA secondary to SLE are usually treated with immunosuppression but less is known about hematopoietic stem cell transplantation (HSCT) as a therapeutic option. We present the case of a 22-year-old Latin-American woman with a known diagnosis of SLE referred to our center due to pancytopenia. She presented joint pain, fatigue, oral ulcers, a non-scarring alopecia, and anemic syndrome. A diagnostic workup revealed severe aplastic anemia (sAA). She received immunosuppressive therapy with corticosteroids, cyclosporine, danazol, and rituximab, which led to a transient partial response. An outpatient HLA-matched related allogeneic HSCT was performed. She received 5.9 x 106 CD34+ cells/kg after conditioning with high-dose cyclophosphamide plus fludarabine and anti-thymocyte globulin. She achieved a complete response and continues in remission with no signs of graft-versus-host disease (GVHD) or SLE activity at 38 months of follow-up. Even though allogeneic hematopoietic stem cell transplantation is not defined as a first-line treatment for severe AA in SLE, the procedure resulted in the complete remission of both related autoimmune diseases in this patient.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease well known for its multisystem involvement.
Hematological manifestations are common, including leukopenia, thrombocytopenia, and autoimmune hemolysis, and they have also been a key component of diagnostic criteria since 1971 [1]. Bone marrow (BM) involvement presenting as aplastic anemia (AA) is rare but can also be present as part of the SLE clinical spectrum [2]. The pathogenesis of AA in the context of SLE has been poorly described. There are reports of an association of both T cell dysregulation [2, 3] and B cell autoantibody production with the capacity to inhibit in vitro BM colony formation from granulocyte-macrophage progenitor cells [2]. Histological findings described in the BM of SLE patients with hematological manifestations at diagnosis include fibrosis, pure red cell, and trilinear aplasia [3].
Patients with AA secondary to SLE are usually treated with immunosuppression, including corticosteroids, cyclosporine (CsA), anti-thymocyte globulin (ATG), or cyclophosphamide, among others [2, 3, 4, 5, 6]. On the other hand, allogeneic hematopoietic stem cell transplantation (HSCT) is considered the best first-line treatment for severe de novo aplastic anemia in patients with an HLA-matched donor [7]. Less is known about HSCT as a therapeutic option for patients presenting with both SLE and AA.
Herein, we report the case of a patient who underwent peripheral blood (PB) allo-HSCT for the treatment of severe AA in the context of SLE. This case report follows the CARE Guidelines [8]. In addition, informed consent was obtained from the patient.
Case Presentation
A 22-year-old Latin-American woman with a previous episode of self-limited infection-related skin vasculitis at age 14 and an otherwise unremarkable history with no family history of rheumatologic, hematological, or autoimmune disease presented with joint pain in her hands, wrists, knees, and ankles (Table 1).
| Year | Symptoms |
| 24-Jul-15 | Onset of symptoms: joint pain in hands, wrists, knees, and ankles. |
| 24-Aug-15 | An episode of menorrhagia and anemic syndrome. She was treated with packed red blood cells, prednisone, and danazol daily by another physician. |
| 31-Aug-15 | The patient was admitted to our hospital for a second opinion. |
| 3-Sep-15 | Establishment of diagnosis in our facility: SLE and sAA. Her previous medication scheme was modified. Rituximab was added since she was transfusion-dependent without immediate access to ATG and eltrombopag. |
| 1-Oct-15 | The patient achieved only a partial response with transfusion independence. |
| 3-Oct-16 | The patient chose to undergo an outpatient allogeneic stem cell transplantation and received a conditioning regimen before transplantation. The infusion was performed without complications in an outpatient basis and received prophylaxis after. |
| 9-Oct-17 | The patient remains in complete remission. |
| 10-Oct-19 | The patient has not developed signs or symptoms of GVHD or SLE. However, she is still positive for ANAs in a 1:160 titer with a fine speckled pattern. |
Table 1: Timeline.
This persisted for a month, followed by an episode of menorrhagia and anemic syndrome. She received five packed red blood cell units and was treated with prednisone 1 mg/kg and danazol 200 mg daily by another physician. One week after this episode, she sought a second opinion at our institution. She complained of fatigue and exertional dyspnea. She had normal vital signs, generalized pallor, nonpainful oral ulcers, and non-scarring alopecia. Synovitis of her wrist and metacarpophalangeal joints were noted. She had no signs of bleeding or active infection. No additional abnormalities were documented in the physical exam. Her laboratory tests revealed a hemoglobin concentration of 5.80 g/dl, a hematocrit of 18.2%, a mean corpuscular volume (MCV), and mean corpuscular hemoglobin (MCH) within normal range, reticulocytes 2.2%, white blood cell (WBC) count of 3.15 x 103/µL with\ 0.311 x 103/µL neutrophils and 13x103/µL platelets. Her blood chemistry panel and liver function tests were unremarkable.
The direct antiglobulin test was negative. BM aspiration was hypocellular, and the biopsy showed 5% cellularity. Further testing for Epstein-Barr virus (EBV), viral hepatitis, cytomegalovirus (CMV), human immunodeficiency virus (HIV), parvovirus B19, and syphilis were negative. Antinuclear antibodies were positive with nucleolar 1:1280, homogeneous 1:640, and fine speckled 1:160 titers; she was also positive for Anti-Ro, anti-DNAds, and lupus anticoagulant. Paroxysmal nocturnal hemoglobinuria was excluded by flow cytometry. Hydroxychloroquine 400 mg/day was added to prednisone, danazol 400 mg/day, and cyclosporine 12 mg/kg/day. As she was transfusion-dependent without immediate access to ATG and eltrombopag, rituximab 100 mg weekly for 4 weeks was added.
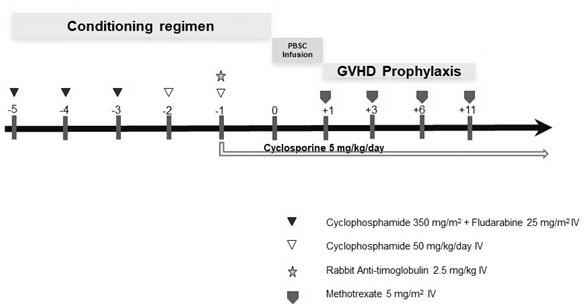
The patient achieved only a partial response with transfusion independence. She had a 6/6 HLA-matched 19-year- old brother as a potential donor and after a shared decision-making process, she chose to undergo an outpatient allogeneic stem cell transplantation (allo-SCT). As a conditioning regimen, she received cyclophosphamide 350 mg/m2 plus fludarabine 25 mg/m2 for three days, followed by two days of cyclophosphamide at 50 mg/ kg/day [9] and, the day before transplantation, 2.5 mg/ kg of rabbit ATG (Figure 1). The infusion of 5.9 x 106 CD34+ cells/kg was performed without complications. Methotrexate and cyclosporine A were given as GVHD prophylaxis. She received broad-spectrum antibiotics due to a suspected urinary tract infection without isolation and received a single platelet transfusion. Neutrophil and platelet engraftment was achieved on day +19 and day +13, respectively. The procedure was performed almost entirely on an outpatient basis, except for the day before transplantation to apply ATG.
After the stem cell infusion, she was discharged. Chimerism on day +100 was reported at 99%. One year after the transplant, she remained in complete remission, the chimerism was 94%, and a follow-up BM biopsy reported a cellularity of 30% with a conserved myelomonocytic lineage, a discrete decrease of erythroid lineage, and a normal megakaryocytic lineage. After 3 years, she has not developed signs or symptoms of GVHD nor SLE with a normal erythrocyte sedimentation rate and CRP; however, she is still positive for ANAs in a 1:160 titer with a fine speckled pattern. Regarding psychosocial aspects, the patient remained optimistic from the beginning of the symptom onset. Some physical limitations were noted on account of the clinical manifestations of anemic syndrome; besides that, she states that no symptoms of depression or anxiety originated from her SLE and sAA diagnosis.
Patient Perspective
The patient currently takes no medication, works a full- time job as an architect, and has no remaining symptoms or complications that could be pronounced as a consequence of the HSCT that she underwent. Ever since the HSCT, the patient repeats routine laboratory tests every 4 to 6 months and they remain between normal ranges, except for the Erythrocyte Sedimentation Rate (ESR) which has been shown to be elevated. Overall, she described the treatment as a process where she felt informed and serene from beginning to end.
Discussion
It is unclear which strategy is best to treat AA in the context of SLE, with immunomodulatory therapies being the preferred approach thus far. A comprehensive literature review reported on the outcome of patients treated with several therapies [10]. The most common drugs used were corticosteroids in 20/26 cases with reported improvement in all, albeit confounded by the use of other therapies. Plasmapheresis, a strategy usually reserved for severe autoimmune phenomena, was used in 6 cases with 4 patients responding. Other immunomodulatory drugs used included cyclosporine, cyclophosphamide, azathioprine, androgens, hydroxychloroquine, intravenous immune globulin, and methotrexate. Of note, rituximab was used by a single patient with a favorable response. Mortality was 15% overall with a short follow-up [10]. On the other hand, the first line of treatment for patients under 50 years of age with sAA with an HLA-identical sibling is allogeneic HSCT [7] with the combination of cyclophosphamide and ATG considered a standard conditioning regimen. In the case described herein, allo-HSCT was offered as curative therapy after only a partial response to immunosuppressive treatment was documented. Considering the overlapping severe diseases that our young patient was diagnosed with, allo- HSCT was considered the best option in this particular case.
Admittedly, HSCT is not considered a standard treatment for SLE, where it is used sparingly in patients with severe or refractory disease. Different mechanisms have been described to demonstrate the efficacy of HSCT. An autologous transplant is intended to “reset” the immune system and in the case of allogeneic HS, replace it [10, 11, 12]. In 2019, a systematic review of HSCT in SLE showed that most studies corresponded to autologous HSCT [13]. A retrospective registry survey carried out by EBMT/EULAR in patients with severe SLE who underwent auto-HSCT reported remission in 33/50 evaluable patients, of which 10/31 subsequently relapsed [11]. A previous study of BM allo-HSCT in 27 patients reported a mean 7.4- month disease-free interval with two deaths [14]. A phase 2 clinical trial in 81 patients studying allogeneic BM and umbilical cord blood-derived mesenchymal stem cells reported a 34% 5-year remission rate, and a post-transplant overall relapse rate in 9/37 (24%) patients that had achieved clinical remission [15]. These studies show that a proportion of patients with severe SLE who undergo HSCT may remain disease-free, although limited by a short follow-up and the known toxicities of transplantation, such as infection and graft-versus-host disease, among others.
To our knowledge, this is the first case using PB as a stem cell source in allogeneic HSCT for AA secondary to SLE besides that, the outpatient procedure is also unique. Previously, an umbilical cord blood transplantation was reported in a 22-year-old woman with sAA related to SLE, she received a conditioning regimen that consisted of fludarabine 30 mg/kg (4 days), cyclophosphamide 30 mg/kg (4 days), ATG 5 mg/ kg divided into two doses, and 2 Gy total body irradiation. Moreover, pre-emptive rituximab was added to prevent Epstein-Barr virus-associated lymphoproliferative disease with a successful outcome after 2 years [10]. In our patient irradiation and rituximab were not administered.
Although BM is considered the best source of stem cells in sAA [7, 16], we have used PBSC successfully in the past [9]. One of the advantages of PBSC transplantation is that it contains 2–3-fold more CD34 cells in contrast to BM; achieving earlier hematological recovery which reduces complications and potentially transplant-related costs and can also be performed on an outpatient basis safely [17]. The association of HSCT and SLE activity makes us question if the source of stem cells affects achieving remission for a relatively long period of 38 months compared to other sources [3, 14, 15]. Even though remission in SLE does not have a universally accepted definition [18], the main goals of treatment are to achieve clinically low or absent disease activity and prevention of exacerbations [19]. Prolonged complete remission in SLE is uncommon [19, 20, 21]. Reaching a prolonged and sustained remission, either with immunomodulatory therapy or HSCT, as well as preventing relapses, remains difficult in these patients.
Our patient has had a favorable clinical evolution for over 3 years without GvHD or other transplantation-related comorbidities, and interestingly, continues to be without SLE activity. Long-term follow-up is mandatory, even if our patient has been in remission, flares have been reported in post-allo-SCT [15].
Conclusion
Allogeneic HSCT can be considered for patients with severe AA and SLE who do not respond adequately to immunosuppressive therapy, considering the potential for disease remission, decreased side effects related to reduced conditioning regimen transplantation, and cost-effectiveness, especially if an outpatient HSCT strategy is chosen. More studies are necessary to evaluate long-term outcomes with the use of PBSC transplantation in severe SLE.
References
-
Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, et al. (2019) European League Against Rheumatism/ American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol 71(9): 1400-1412.
-
Anderson E, Shah B, Davidson A, Furie R (2018) Lessons learned from bone marrow failure in systemic lupus erythematosus: Case reports and review of the literature. Semin Arthritis Rheum 48(1): 90-104.
-
Chalayer E, Costedoat-Chalumeau N, Beyne-Rauzy O, Ninet J, Durupt S, et al. (2017) Bone marrow involvement in systemic lupus erythematosus. Qjm 110(11): 701-711.
-
Hinterberger-Fischer M, Hocker P, Lechner K, Seewann H, Hinterberger W (1989) Oral cyclosporin-A is effective treatment for untreated and also for previously immunosuppressed patients with severe bone marrow failure. Eur J Haematol 43(2): 136-142.
-
Walport MJ, Hubbard WN, Hughes GRV (1982) Reversal of aplastic anaemia secondary to systemic lupus erythematosus by high-dose intravenous cyclophosphamide. Br Med J 285(6344): 769-770.
-
Liu W, Hu Z, Lin S, He J, Zhou Y (2014) Systemic lupus erythematosis with severe aplastic anemia successfully treated with rituximab and antithymocyte globulin. Pak J Med Sci 30(2): 449-451.
-
Bacigalupo A (2017) How I treat acquired aplastic anemia. Blood 129(11): 1428-1436.
-
Riley DS, Barber MS, Kienle GS, Aronson JK, Schoen- Angerer TV, et al. (2017) CARE Explanation and Elaborations: Reporting Guidelines for Case Reports. J Clin Epi (89): 218-235.
-
Gomez-Almaguer D, Vela-Ojeda J, Jaime-Perez JC, Gutierrez-Aguirre CH, Cantu-Rodríguez OG, et al. (2006) Allografting in patients with severe, refractory aplastic anemia using peripheral blood stem cells and a fludarabine- based conditioning regimen: The Mexican experience. Am J Hematol 81(3): 157-161.
-
Chalayer E, Ffrench M, Cathebras P (2015) Aplastic anemia as a feature of systemic lupus erythematosus: a case report and literature review. Rheumatol Int 35(6): 1073-1082.
-
Jayne D, Passweg J, Marmont A, Farge D, Zhao X, et al. (2004) Autologous stem cell transplantation for systemic lupus erythematosus. Lupus 13(3): 168-176.
-
Alchi B, Jayne D, Labopin M, Kotova O, Sergeevicheva V, et al. (2013) Autologous haematopoietic stem cell transplantation for systemic lupus erythematosus: Data from the European Group for Blood and Marrow Transplantation registry. Lupus 22(3): 245-253.
-
De Silva NL, Seneviratne SL (2019) Haemopoietic stem cell transplantation in Systemic lupus erythematosus: A systematic review. Allergy, Asthma Clin Immunol 15: 1-12.
-
Vanikar AV, Modi PR, Patel RD, Kanodia KV, Shah VR, et al. (2007) Hematopoietic Stem Cell Transplantation in Autoimmune Diseases: The Ahmedabad Experience. Transplant Proc 39(3): 703-708.
-
Wang D, Zhang H, Liang J, Wang H, Hua B, et al. (2018) A Long-Term Follow-Up Study of Allogeneic Mesenchymal Stem/Stromal Cell Transplantation in Patients with Drug-Resistant Systemic Lupus Erythematosus. Stem Cell Reports 10(3): 933-941.
-
Bacigalupo A, Socie G, Schrezenmeier H, Tichelli A, Locasciulli A, et al. (2012) Bone marrow versus peripheral blood as the stem cell source for sibling transplants in acquired aplastic anemia: Survival advantage for bone marrow in all age groups. Haematologica 97(8): 1142- 1148.
-
Ruiz-Arguelles GJ, Gomez-Almaguer D, Ruiz-Arguelles A, Gonzalez-Llano O, Cant OG (2001) Results of an outpatient-based stem cell allotransplant program using nonmyeloablative conditioning regimens. Am J Hematol 66(4): 241-244.
-
Van Vollenhoven R, Voskuyl A, Bertsias G, Aranow C, Aringer M, et al. (2017) A framework for remission in SLE: Consensus findings from a large international task force on definitions of remission in SLE (DORIS). Ann Rheum Dis 76(3): 554-561.
-
Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, et al. (2019) 2019 Update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis 78(6): 736-745.
-
Urowitz MB, Feletar M, Bruce IN, Ibañez D, Gladman DD (2005) Prolonged remission in systemic lupus erythematosus. J Rheumatol 32(8): 1467-1472.
-
Medina-Quinones CV, Ramos-Merino L, Ruiz-Sada P, Isenberg D (2016) Analysis of Complete Remission in Systemic Lupus Erythematosus Patients Over a 32-Year Period. Arthritis Care Res 68(7): 981-987.
- Psychogenic Erectile Dysfunction in Late Adulthood: A Case Report on Clinical Intervention and Intimacy Restoration
- Clinical Trials on COVID-19 in 2025: A New Chapter in Global Health Research
- Innovations and Challenges in Contemporary Medical Clinical Trials: An Editorial Perspective
- Innovations and Challenges in Contemporary Medical Clinical Trials: A Critical Perspective
- Reimagining Clinical Trials: The Power of Continuous Feedback from Medical Reports
- Factors Influencing Brain Drain: Perspectives from a Medical School in Turkey