A Case of Late Presenting Retinitis Pigmentosa in an Adult Female
This study aimed to bring to light a case of non-syndromic Retinitis Pigmentosa in a middle-aged female. A 39-year-old female complained of blurred vision that started 3 years ago. It initially affected her distant vision, mainly in low-light conditions, followed by the involvement of near vision as well. On fundus examination, bony spicule-like pigmentation was found peripherally, waxy pallor of the optic disc was present, severe arteriolar attenuation was found, and macular edema was present. The patient was prescribed vitamin A 15,000 IU and asked to come for a follow-up for a low visual aid assessment. This case report highlights the devastating effects of Retinitis Pigmentosa and the need for early diagnosis to slow the progression of vision loss. The peculiarity of this report lies in the fact that the patient showed symptoms very late, when usually RP is diagnosed in adolescence which also progressed rapidly within 3 years to involve distant and far vision. The absence ofany family history is also notable as it may indicate an autosomal recessive inheritance with low-penetrance or a sporadicmutation.
Vongole H¹, Onkar A²*, Kumari R³ and Gupta M⁴
¹MBBS 3rd Professor, All India Institute of Medical Sciences (AIIMS), India ²Associate Professor, Department of Ophthalmology, All India Institute of Medical Sciences (AIIMS), India ³Assistant Professor, All India Institute of Medical Sciences (AIIMS), India ⁴MBBS, PG Resident 2nd year, All India Institute of Medical Sciences (AIIMS), India
Abbreviations
RP: Retinitis Pigmentosa; arRP: Autosomal Recessive RP; adRP: Autosomal Dominant RP; xlRP: X-linked RP.
Introduction
Retinitis Pigmentosa (RP) is a group of genetic eye disorders characterized by the progressive degeneration of photoreceptor cells in the retina, leading to vision loss [1]. The term “Retinitis Pigmentosa” is a misnomer, as the name might suggest an inflammatory etiology that is not the primary cause of the disease [2]. Non-syndromic RP has a worldwide prevalence of 1: 4000 [3], with prevalence as high as 1:930 in urban, and 1:372 in rural populations of South India [4], hence it is a cause of major concern, especially in India. Non-syndromic RP may be inherited as autosomal dominant RP(adRP) which contributes to 15%-25% of all RP, autosomal recessive RP (arRP) which contributes to 5%-20% of all RP, X-linked RP (xlRP) contributing to 5%-15% of all RP
and digenic forms which are very rare [5]. Men are slightly more affected than women due to the X-linked form being expressed more in men [1]. RP is usually bilateral however there have been some reports of unilateral RP [6]. The typical presentation of RP involves complaints of visual disturbances beginning at around 20 years [1]. The visual disturbance may include night blindness, followed by concentric visual field loss, due to rod dysfunction. Central vision loss occurs later in life due to cone dysfunction [3]. Physical examination on fundoscopy often reveals the triad of optic disc pallor, bony spicule pigmentation, and attenuation of retinal arterioles [7].
Retinitis Pigmentosa may be diagnosed by genetic testing, electroretinography, visual field testing, and optical coherence tomography. There is no single treatment for RP because over 100 genes cause it [8]. The genes involved in causing RP include ABCA4, BEST1, PDE6A, PDE6B, and many more [9].
Clinical Presentation
History: A 39-year-old female presented to the ophthalmology outpatient department with chief complaints of blurred vision that started 3 years ago. Her distant vision was initially affected, and objects appeared hazy in low-light conditions. This then progressed to involve her near vision as well. There was no similar complaint in the family. She has no known comorbidities. No other systemic complaints were present.
Examination: Her visual acuity was found to be 6/60 in both eyes. Her IOP was 17.1 mmHg in the right eye and 14.4 mmHg in the left eye. On fundus examination, bony spicule- like pigmentation was found peripherally, waxy pallor of the optic disc was present, severe arteriolar attenuation was found, and macular edema was present (Figures 1A & 1B). On slit-lamp examination, a posterior subcapsular cataract was found.
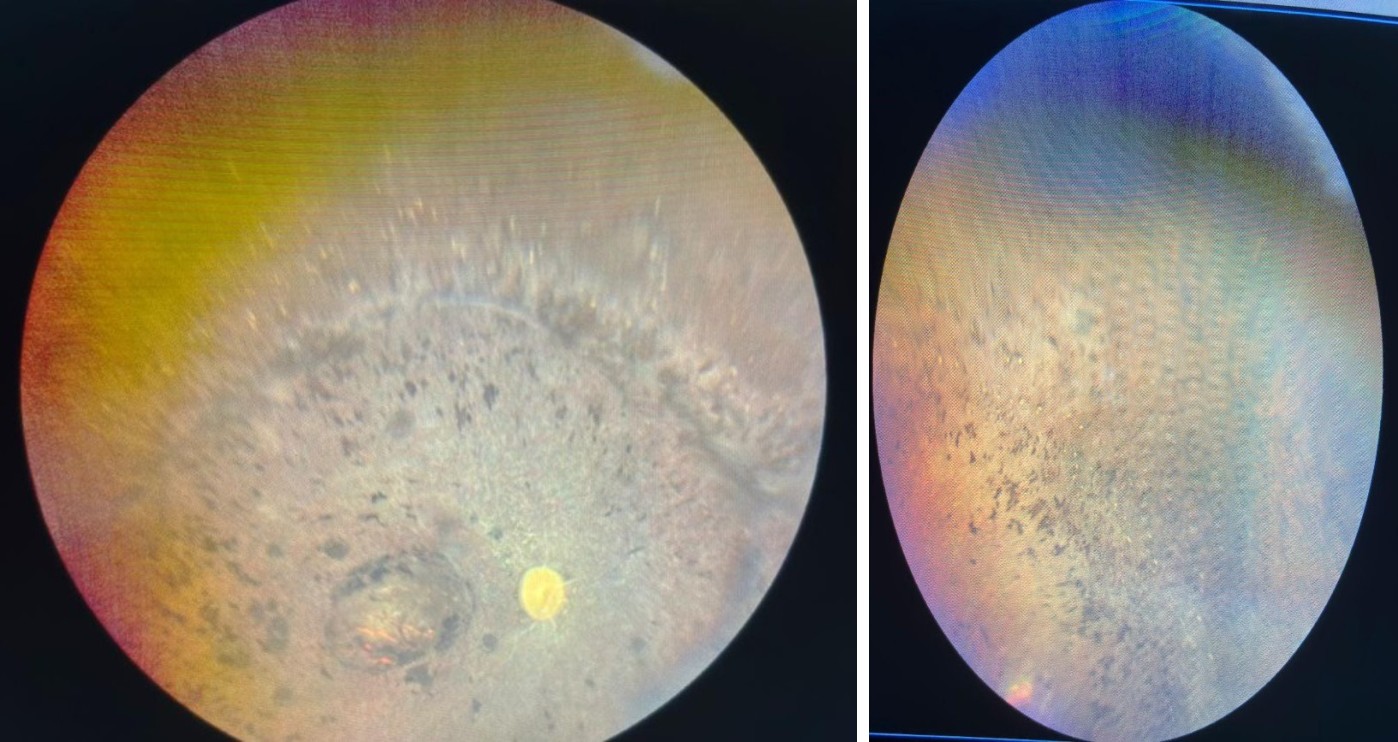
Figure 1A & 1B: 1A: Fundus picture of Right eye showing waxy pale disc, arteriolar attenuation and peripheral bony spicule like pigmentation. 1B: Peripheral fundus picture of left eye showing the characteristic pigmentation.
Based on history and characteristic fundus findings, a presumptive diagnosis of Retinitis Pigmentosa was made. Routine blood investigations, including a complete blood count, were taken to rule out acanthocytes. Differential Diagnosis:
- Cone rod dystrophy
- Traumatic Retinopathy
- Drug-induced Retinopathy _
- _Retinitis Pigmentosa associated syndromes
- Inflammatory/Infective Retinopathy Traumatic and drug-induced retinopathy were ruled out due to the absence of a history of trauma or intake of retino- toxic drugs respectively. Syndromic RP was ruled out by the absence of systemic features, and Ushers syndrome the most common syndrome associated with RP was ruled out by the absence of sensorineural hearing loss. Inflammatory retinopathy was ruled out by the absence of signs of inflammation such as photosensitivity and pain.
Management: The patient was prescribed vitamin A 15000 IU and asked to come for a follow-up for a low vision aid assessment [10]. She was referred to the ENT department to rule out hearing loss associated with Usher syndrome. The patient was also told to have her immediate family and children screened for Retinitis Pigmentosa, as it may be transmitted in an autosomal dominant or autosomal recessive pattern.
Discussion: This case highlights the progressive nature of Retinitis Pigmentosa (RP) and its significant impact on vision. RP as a hereditary condition leads to the degeneration of photoreceptors by apoptosis, mainly affecting the rods followed by the eventual involvement of cones, which in turn explains the initial symptom loss of vision dim-light conditions as shown by the patient’s history. There is also presence of pigmentary clumps in the retina due to migration from retinal pigment epithelium, earning the disease its name. The patient’s clinical presentation, characterized by blurred vision, bony-spicule pigmentation, optic disc pallor, and macular edema, is consistent with the classic findings of RP. No systemic abnormalities were present, suggesting this is a non-syndromic variant of Retinitis Pigmentosa. There was also an absence of any family history, suggesting that it may indicate an autosomal recessive inheritance with low penetrance or a sporadic mutation. This patient had a late- onset of symptoms with a rapid progression which made this case peculiar and different from the classical progression of RP. This eventually led her to present at an advanced stage with severe visual impairment affecting her daily life.
There being no definitive treatment for RP made management difficult however a study showed that vitamin A therapy of 15,000 IU/day slowed down vision loss over 5 years for patients with RP [10].
Another study however found that there were no clear benefits of treatment with vitamin A for people with RP [11]. Hence, more research is needed to determine if vitamin A is a viable option for the management of RP. Taking these findings into consideration, the patient was prescribed vitamin A 15,000 IU/day and was asked to follow-up. She was also referred to the ENT department to rule out Usher syndrome, the most common syndromic variant of RP representing 18% of all RP cases [12] and was recommended to get her family screened for RP which add a preventive care aspect that is not always emphasized in RP cases. Early diagnosis through clinical examination and appropriate diagnostic testing is crucial as it allows for timely management, this however was not possible in this case due to the advanced stage of disease at presentation. Retinitis Pigmentosa being a complex, multi-gene disorder has no single treatment available. The most advanced options are the Argus retinal prosthesis and stem cell therapy, which are highly expensive and often inaccessible to the average person.
References
-
O’Neal BT, Tripathy K, Luther EE (2024) _Retinitis_ _Pigmentosa_. StatPearls, NCBI Bookshelf.
-
Thenappan A, Nanda A, Lee CS, Lee SY (2023) _Retinitis_ _Pigmentosa_ Masquerades: Case Series and Review of the Literature. Journal of Clinical Medicine 12(17): 5620.
-
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, et al. (2018) Non-syndromic _Retinitis_ _Pigmentosa_. Prog Retin Eye Res 66: 157-186.
-
Sen P, Bhargava A, George R, Ramesh SV, Hemamalini A, et al. (2008) Prevalence of _Retinitis Pigmentosa_ in South Indian population aged above 40 years. Ophthalmic Epidemiol 15(4): 279-281.
-
Fahim AT, Daiger SP, Weleber RG (1993) Nonsyndromic _Retinitis Pigmentosa_ Overview. In: Adam MP, Feldman J, et al. (Eds.), GeneReviews®. University of Washington, USA.
-
Weller JM, Michelson G, Juenemann AG (2014) Unilateral _Retinitis Pigmentosa_: 30-year follow-up. BMJ Case Reports. 2014.
-
Tripathy K, Sharma YR, Chawla R, Basu K, Vohra R, et al. (2017) Triads in Ophthalmology: A Comprehensive Review. Semin Ophthalmol 32(2): 237-250.
-
Boyd K (2024) What Is _Retinitis Pigmentosa_. American Academy of Ophthalmology.
-
Daiger S, Sullivan L, Bowne S (2013) Genes and mutations causing _Retinitis Pigmentosa_. Clin Genet 84(2): 132-141.
-
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, et al. (1993) A Randomized Trial of Vitamin A and Vitamin E Supplementation for _Retinitis Pigmentosa_. Arch Ophthalmol 111(6): 761-772.
-
Rayapudi S, Schwartz SG, Wang X, Chavis P (2013) Vitamin A and fish oils for _Retinitis Pigmentosa_. Cochrane Database Syst Rev 2013(12): 33.
-
Toms M, Pagarkar W, Moosajee M (2020) Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Therapeutic Advances in Ophthalmology 12: 2515841420952194.
- Screening of Hospital Staff During World Glaucoma Week in a Tertiary Eye Care Centre
- Angioid Streaks with Macular Neovascularization: Clinical Insights from Two Cases
- Giant Kissing Naevus: An Oculoplastic Challenge
- Why Freedom of Vision Should Not Cost the Freedom of Feeling - LASIK in the Climate of Change
- Asymmetric Optic Nerve with Small Disc and Large Cup: A Rare and Challenging Case of Unilateral Optic Nerve Hypoplasia
- Large Angle Exotropia in a Child: A Case Report