Atypical Hemolytic Uremic Syndrome: Atypical Course and Atypical Mutations Combination
Atypical hemolytic uremic syndrome (aHUS) is a rare thrombotic microangiopathy, associated with dysregulation of the alternative pathway of the complement system, characterized by hemolytic anemia, thrombocytopenia, and acute renal failure. In this report, we discuss the case of a woman with aHUS that carries heterozygous C3, complement factor H and membrane cofactor protein mutations, with an uncommon presentation, clinical course and outcome.
António C1
Abel Salazar 4099-001 Porto, Portugal
Coimbra, Portugal
Portugal, Telephone: +351 222 077 500; Fax: +351222033189; Email: soacorreia@gmail.com Membrane cofactor protein
Introduction
Atypical hemolytic uremic syndrome (aHUS) pathogenesis is strongly associated with complement dysregulation [1, 2]. HUS is a thrombotic microangiopathy (TMA) clinically defined by microangiopathic hemolytic anemia, thrombocytopenia and acute renal failure. Mutations and polymorphisms in genes encoding regulatory proteins such as complement factor H (CFH), complement factor I (CFI) and membrane cofactor protein (MCP/CD46), and the complement activating components C3 and factor B (CFB) have been identified. Atypical hemolytic uremic syndrome associated mutations in regulators are loss-of-function, while mutations in activators are gain-of-function [2, 3]. Penetrance of the disease is 50%, which explain that the age at onset and severity of the disease may vary among family members [2]. It is still unclear why aHUS has incomplete penetrance among the mutation carriers. Eculizumab is a recombinant, humanized, monoclonal immunoglobulin G antibody that targets C5 and has reduced the magnitude of the thrombotic microangiopathy, restored kidney function and improved quality of life but, however, is expensive [2, 4, 5]. In this report, we discuss the case of a patient with aHUS that carries a pathogenic C3 mutation combined with a CD46 variant and the risk haplotypes MCPggaac and CFH-H3, with an atypical presentation, clinical course and outcome.
Case Report
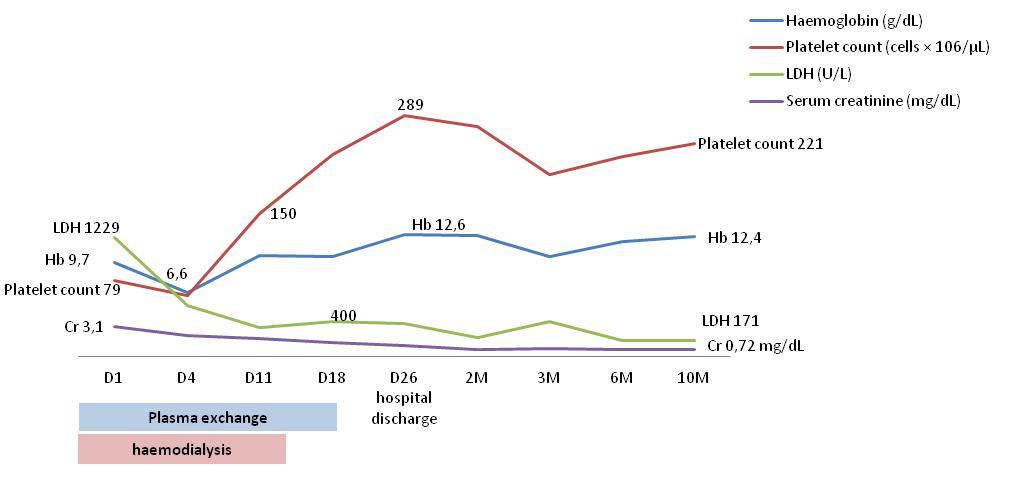
A 20-year-old female presented in our hospital with abdominal pain with no previous history of diarrhea or fever. She had one uneventful pregnancy at the age nineteen and no other relevant past medical history. She had no family history of kidney disease or aHUS. On examination she had a blood pressure of 118/56 mmHg, pulse of 60, respiratory rate of 18, oliguric and temperature of 36.5ºC. On auscultation of lungs and heart was normal. There was no jugular venous distention. There was any skin rash or purpura. In the abdominal examination there was no distension, rigidity or rebound tenderness, but with generalized pain with deep palpation. Neurological exam result was normal. The initial investigation revealed: Hemoglobin 9.5 g/L normocytic normochromic, reticulocytosis of 1.40%, platelet count 79 × 106/μL and serum creatinine 3.1 mg/dL accompanied by hematuria (>25 per high-power field) and proteinuria (400 mg/dL). A peripheral blood smear revealed schistocytes, lactate dehydrogenase (LDH) level was elevated to 1229U/L and haptoglobin was <0.243 g/L. Direct Coombs test was negative. Renal Ultrasound showed normal renal size, normal parenchyma-sinus differentiation and excluded obstruction. Abdominal ultrasound documented cholecystitis, which was assumed to be ischemic as a consequence of TMA. Her complement levels (reference ranges) were as follows: Low C3, 43.9mg/dL (81–167mg/dL); normal C4, 30.2mg/dL (11–42mg/dL); CFB 44.8 mg/dL (19.1 - 38.2) and CHF 73.7 mg/dl (34.5 – 59.0). She had a negative ANA and anti-DNA assays. Stool culture results were negative for E. coli O157. ADAMTS13 activity levels, prior to PE initiation, were 101% (40-130%); CH50, 92.1% (69- 129%) and AH50, 65.8% (30-113%). Due to the clinical presentation with microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury with oliguria supported by hemodialysis, suspicion was high for atypical hemolytic-uremic syndrome and plasma exchange (PE) was started. Following ten sessions of PE over a period of 11 days, her parameters improved (haemoglobin 10.5 g/dL, platelet count 148× 106/μL, LDH 229U/L) and she recovered renal function after 9 days of haemodialysis (Table 1 and Figure 1).
| Hospital Admission Hospital | |||||||||||||
| Follow Up | |||||||||||||
| Discharge | |||||||||||||
| D1 | D4 | D11 | D18 | D26 | 2 months | 3 months | 6 months | 10 months | |||||
| Hemoglobin (g/ dL) | 9,7 | 6,6 | 10,5 | 10,4 | 12,6 | 12,5 | 10,4 | 11,9 | 12,4 | ||||
| Platelet count (cells × 106/μL) | 79 | 63 | 148 | 209 | 250 | 238 | 188 | 207 | 221 | ||||
| LDH (U/L) | 1229 | 525 | 297 | 366 | 339 | 199 | 367 | 169 | 171 | ||||
| Serum creatinine (mg/dL) | 3,1 | 2,18 | 1,87 | 1,44 | 1,16 | 0,74 | 0,84 | 0,75 | 1,72 | ||||
| Complement C3 (mg/dL) | 43,9 | 51,9 | 87,4 | 51,4 | |||||||||
| Complement C4 (mg/dL) | 30,2 | 25,9 | 48,8 | 35,9 |
Table 1: Biochemical parameters variations. C3 normal range: 81–167mg/dL; C4 normal range: 11–42mg/dL.
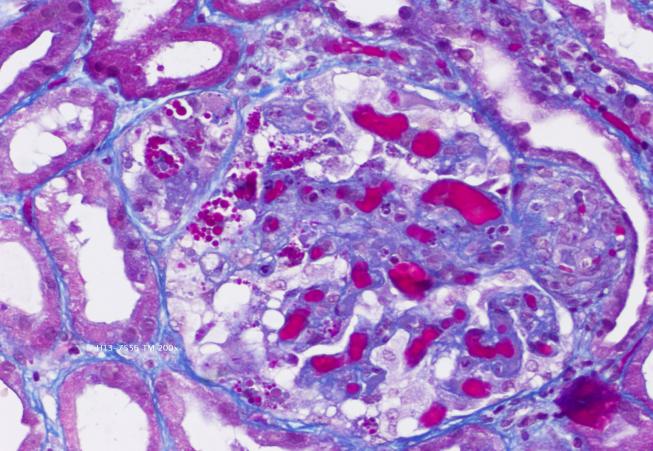
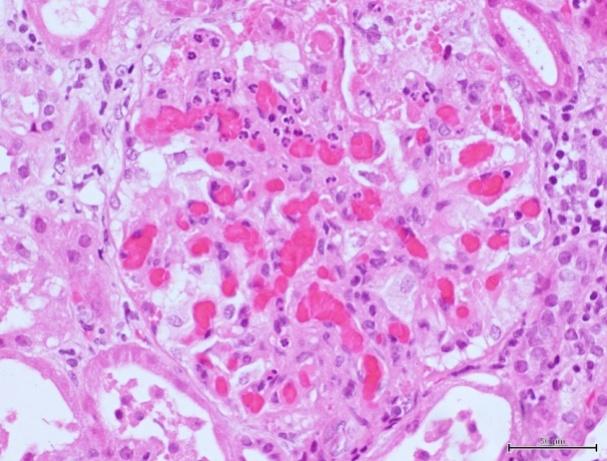
Masson's trichrome stain (200x) showing afferent arterioles with fibrinoid necrosis and fragmented erythrocytes (on the left) and hematoxylin and eosin stain with congestion filling up capillary loops and mesangiolysis (on the right).
After 10 months of follow up she is clinically stable and without disease relapses. The molecular studies of complement genes were performed by a next generation sequencing (NGS) - based gene panel (ADAMTS13, CFH, CFHR1, CFHR3, CFHR4, CFHR5, CFI, CFB, C3, THBD and DGKE) using Ion Hi-Q™ Sequencing 200 Kit chemistry on an Ion Torrent™ PGM sequencing system (Thermo Fisher Scientific).
As recommended by the practice guidelines for the evaluation of pathogenicity recently published by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [6]. i) We checked the frequency of variants found in the Exome Aggregation Consortium (ExAC), Exome Variant Server (EVS), Human Gene Mutation Database and FH aHUS Mutation Database
ii) Variants were classified as pathogenic, likely pathogenic, uncertain significance, likely benign or benign based on the available evidence [6]. This molecular study approach found in this patient a C3-combined variant: A pathogenic missense mutation in C3, c.1775G>A, p.Arg592Gln; a benign missense variant of CD46, c.686G>A, p.Arg229Gln; and also had the risk haplotypes MCPggaac and CFH-H3. All variants were in the heterozygous state. The benign variant CD46 p.Arg229G in was only found in a population database (MAF: ExAC-EA, 0.0063) and therefore has an uncertain significance. After 10 months follow up she is clinically stable and without disease relapses. Despite having infectious complications (tonsillitis with high fever) there was no recurrence of HUS. C3 levels remained persistently low. Given her clinical stability after a good response to plasmapheresis and without disease relapse, the patient hasn’t started eculizumab.
Discussion
Mutations in C3 gene were first associated with aHUS in 2008 by Frémeaux-Bacchi and colleagues, including the complement C3_p.Arg592Gln mutation [7]. Since then, other mutations are being found [8]. _C3 mutations cause a defect of the ability of C3 to bind to regulatory protein MCP and are an indirect gain of function mutation leading to an increased capacity for CFB to bind to C3b and an increased formation of the C3 convertase. Converge at the cleavage of the terminal complement protein C5 leading to endothelial injury, and finally thrombotic microangiopathy. Plasma C3 levels are low in about 70% of patients that correlated with disease severity [8]. Lhotta, et al. suggests that, apart from aHUS, p.Arg592Gln mutation can be related with hypertension, haematuria and chronic kidney [9]. This pathogenic C3 mutation was located near the C3/CFH binding sites, CFH- SCR3 and CFH-SCR4, affecting interactions with CFH [8]. In addition, this study has shown that p.Arg592Gln as the variants p.Arg161Trp and p.Arg592Trp were associated with decreased MCP cofactor activity [8].The worst prognosis is associated with CFH or C3_mutations, with mortality rates or progression to chronic kidney disease during the first year following the initial event of approximately 50-70% (the risk associated with _CD46_mutation is 0-6%)[2, 4]. Plasma treatment could remove mutant _C3 and provide regulatory plasma proteins to act against complement activation induced by mutant C3 [10].
Richards et al in 2003 were the first to report mutations of CD46 [11]. The loss of function mutation of CD46 leads to a low C3b-binding and cofactor activity. The haplotypes MCPggaac and CFH-H3 are the most relevant polymorphisms associated with aHUS [4]. Clinical evolution varies widely depending on the patient's mutation; the one related to the best prognosis is the CD46 mutation [2]. C3 levels in CD46-mutated patients are most often normal. If C3 levels are low, it is likely that another mutation responsible for the activation of complement in the fluid phase is present. As MCP is not a circulating protein, a beneficial effect of plasmatherapy is unlikely to be expected in these patients. Of note, at least ninety per cent of patients undergo remission from acute episodes, whether or not they receive plasmatherapy [2, 12]. CFH mutations were the first identified, more than 200 different mutations are described. Must mutations are heterozygous, but homozygous mutations can be observed and are typically associated with very low C3 and CFH plasma concentrations [2]. Patients with mutation in CD46 and CFI frequently had a second mutation in other complement genes. Patients with CFH, C3 or CFB mutations are less frequently associated with other mutations. This observed fact can suggest that these mutations alone may be sufficient to cause aHUS[13]. The concurrence of C3 mutations with risk polymorphisms in the CFH and CD46 genes modulates penetrance and clinical severity of disease in aHUS [3, 13]. Our patient clinical course was clearly distinct from patients with other C3_p.Arg592Gln or _CFH-H3 mutations and was similar to patients with CD46 mutations described previously. We can induce that even when an unfavorable group of risk factors co-segregates, the disease may not manifest in childhood. A trigger is necessary to initiate the disease, in 50-90% aHUS is preceded by infection and the presence of protective or risk polymorphisms will influence the progression of the disease. To the best of our knowledge, only two cases with C3 mutations have been reported, with favorable long term outcome [3, 14]. Eculizumab is a monoclonal antibody that inhibits the activation of C5 and the formation of the cell membrane attack complex, which is responsible for the damage produced to native cell structures in aHUS. In prospective studies, eculizumab effectively interrupted the process of TMA, associated with long-term favorable outcomes, being know accept as a first line of treatment in aHUS [2, 4, 5]. Our patient was and still is a challenging case, because it was not clear what the triggering event was, she did not manifest the disease when she was exposed to possible triggers (pregnancy, infection), and presented extra-renal involvement (ischemic acute cholecystitis). Our patient illustrates the complexity of aHUS, which depends on multiple predisposing factors: gene mutations, polymorphisms and environmental precipitating factors. In the future, better understanding of the pathophysiology will have implications in the diagnosis, treatment and prognosis of these patients.
References
-
Noris M, Remuzzi G (2009) Atypical Hemolytic– Uremic Syndrome. N Engl J Med 361(17): 1676-1687.
-
Loirat C, Frémeaux-bacchi V (2011) Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 8(6): 60
-
Martínez-barricarte R, Heurich M, López-perrote A, Tortajada A, Pinto S, et al. (2015) The molecular and structural bases for the association of complement C3 mutations with atypical hemolytic uremic syndrome. Mol Immunol 66(2): 263-273.
-
Campistol JM, Arias M, Ariceta G, Miguel Blasco, Mario Espinosa, et al. (2012) Actualización en síndromehemolíticourémicoatípico : diagnóstico y tratamiento . Nefrologia 33(1): 11781.
-
Licht C, Greenbaum LA, Muus P, Babu S, Bedrosian CL, et al. (2015) Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 87(5): 1061- 1073.
-
Richards S, Aziz N, Bale S, Bick D, Das S, et al. (2015) Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5): 405-424.
-
Fre V, Miller EC, Liszewski MK, Strain L, Blouin J, et al. (2008) Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 112(13): 4948-4952.
-
Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, et al. (2015) Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood 125(15): 2359-2369.
-
Lhotta K, Janecke AR, Scheiring J, Petzlberger B, Giner T, et al. (2009) A Large Family with a Gain - of - Function Mutation of Complement C3 Predisposing to Atypical Hemolytic Uremic Syndrome, Microhematuria, Hypertension and Chronic Renal Failure. Clin J Am Soc Nephrol 4(8):1356-1362.
-
Noris M, Caprioli J, Bresin E, Chiara M, Gaia P, et al. (2010) Relative Role of Genetic Complement Abnormalities in Sporadic and Familial aHUS and Their Impact on Clinical Phenotype. Clin J Am Soc Nephrol 5(10):1844-1859.
-
Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, et al. (2003) Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci U S A 100(22):12966-12971.
-
Caprioli J, Noris M, Brioschi S, Gaia Pianetti, Federica Castelletti, et al. (2006) Genetics of HUS : the impact of MCP,CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108(4): 1267-1279.
-
Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, et al. (2013) Combined Complement Gene Mutations in Atypical Hemolytic Uremic Syndrome In fluence Clinical Phenotype. J Am SocNephrol 24(3): 475-486.
-
Siomou E, Gkoutsias A, Serbis A, Kollios K, Chaliasos N, et al. (2016) aHUS associated with C3 gene mutation : a case with numerous relapses and favorable 20-year outcome. Pediatr Nephrol 31(3): 513-517.
- Results of 6-Month Follow-Up of Patients After B-Turp and Thulep
- The Effect of Drinking Water with a High Content of Antimony and Arsenic on the Dynamics of their Distribution in the Kidneys and the Renal Excretory Function in Rats
- Effectiveness and Safety of Tansurethral Thulium Laser Enucleation of the Prostate in the Treatment of BPH: Review
- A Systematic Review on Molecular Pathophysiology Involved in Chronic Kidney Disease and the Role of Animal Models in Drug Discovery to Manage in Chronic Kidney Disease - An Update
- Functional Development of Kidneys in Human Ontogenesis
- Testicular Metastasis: Uncommon Prostate Cancer Case Report