Shiga Toxin-Producing Escherichia Coli Associated Hemolytic Uremic Syndrome
Shiga toxin-producing Escherichia coli (STEC)-associated hemolytic uremic syndrome (STEC-HUS) is a clinical syndrome involving hemolytic anemia (with fragmented red blood cells), low levels of platelets in the blood (thrombocytopenia), and acute kidney injury (AKI). It is the major infectious cause of AKI in children. Severe forms can be associated with multiorgan involvement during the acute stage of the disease. Endothelial injury is the trigger event in the microangiopathic process. The host inflammatory response to toxin and E. coli lipopolysaccharide (LPS) is involved in disease pathophysiology. Early diagnosis and identification of underlying pathogenic mechanisms are of great significance for improving prognosis and reducing sequelae and mortality. Typical management of STEC-HUS patients relies on supportive care for electrolyte and water imbalance, anemia, hypertension, and renal failure. At present there is no specific therapy to ameliorate the prognosis. The Immediate outcome is most often favorable but long-term renal sequelae are frequent due to nephron loss. This review summarizes current knowledge regarding the epidemiological findings, the pathophysiological and clinical aspects, and its diagnosis and management.
Introduction
Shiga toxin-producing Escherichia coli (STEC)- associated hemolytic uremic syndrome (STEC- HUS) is a clinical syndrome characterized by hemolytic anemia (with fragmented red blood cells), low levels of platelets in the blood (thrombocytopenia), and acute kidney injury (AKI) [1]. HUS is a microvascular occlusive disorder that belongs to the category of diseases known as thrombotic microangiopathies (TMAs) [2]. It is a systemic disorder in which severe forms can be associated with multiorgan involvement during the acute stage of the disease. HUS is broadly classified as typical (or post-diarrheal or diarrhea-positive D+) and atypical [3, 4]. Shiga toxin-producing E. coli is also called “typical” HUS, as opposed to “atypical” HUS, which results from alternative complement pathway dysregulation, and “secondary” HUS, caused by various co-existing conditions [4, 5]. Typical HUS is associated with infections by Shiga-like toxin-(Stx) producing bacteria, such as enterohemorrhagic E. coli (EHEC), more appropriately referred to as STEC-HUS. Approximately 5% of HUS cases in children are not associated with Stx-producing bacteria and result from infection by neuraminidase- producing Streptococcus pneumoniae, these forms classified as pneumococcal- HUS (or neuraminidase-associated HUS) [6].
During the last two decades, genetic or acquired defects leading to the dysregulation of the complement system, an important effector mechanism of the innate immune system, have been discovered in about 60 % of patients with atypical HUS (aHUS) [7]. Other causes of aHUS are recessive forms associated with cobalamin C and diacylglycerol kinase epsilon and DGKE and WT1- associated HUS [7].
Secondary HUS may occur as a complication of autoimmune diseases, pregnancy, malignant hypertension, and transplantation [8]. These forms are called secondary HUS, however many of the above conditions often act as triggers of the disease in individuals with a genetic background leading to complement dysregulation [9].
This review summarizes the epidemiological findings and the pathological and clinical aspects of STEC-HUS. Supportive therapy and potential therapeutic strategies are also discussed.
Epidemiology
The annual incidence of STEC-HUS is about 2/100,000 in adults and 6.1/100,000 in children under the age of 5 years [10]. It may be sporadic or present as an outbreak. STEC- HUS is widely distributed throughout the world. In contrast, in Argentina, STEC-HUS is endemic, and approximately 400 new cases are reported annually. During the last decade, the annual incidence has ranged from 8 to 10 cases per 100,000 children under 5 years of age and the case fatality rate remains between 1 and 4% [11]. In Argentina, STEC- HUS is the main cause of AKI in children and the second most common cause of chronic kidney failure, accounting for 20 % of renal transplants in children and adolescents [12]. Consequently, HUS implies large economic costs for the health system.
Most cases are sporadic, but outbreaks are often reported due to a common contaminated food or water source. Cattle constitute the main reservoir, although Shiga toxin-producing E. coli (STEC) strains are usually part of the microbiota of three ruminant species: cattle, pigs, and goats. The high prevalence in calves suggests that they are exposed to the toxin from birth and could indicate an important role in the vertical transmission of these pathogens [13].
Infection in humans occurs following ingestion of contaminated undercooked meat, unpasteurized milk or milk products, water, fruits, or vegetables. Secondary human-to- human transmission is also a major risk factor for HUS [13] and may be a concern in daycare centers.
STEC isolates belong to a large number of O:H serotypes, but STEC O157:H7 is the most prevalent serotype associated with severe human diseases [11]. The predominant STEC serotype causing HUS is O157:H7, responsible for around 2000 such cases annually worldwide. However, non-O157 serotypes have been increasingly reported to account for HUS cases [13]. The major pathogenic E. coli O104:H4 (STEC O194:H7) outbreak that occurred in central Europe during late spring of 2011 highlighted that the pathogenesis of HUS is incompletely understood. More than 4,000 persons were infected mainly in Germany, and it produced more than 900 cases of HUS resulting in 54 deaths [14].
The bacteria are only present in the stools for a few days and, even if present, may not be detected by culture from stool samples. In the first six days after onset of diarrhea, the rate of stool isolation is substantially higher. Only 10–15% of children with E. coli O157 colitis eventually develop HUS [15] suggesting that, in addition to pathogen factors, host factors also contribute to its development.
Pathophysiology of STEC-HUS
Shiga-Toxin Production
STEC produces two main types of Shiga toxins (Stx) known as Stx1 and Stx2, encoded by the stx1 and stx2 genes. Stx is composed of a single A subunit (30 kDa) and five B subunits (each 7 kDa); the former is biologically active and the latter binds to the cell surface receptor. The A subunit (Stx A) possesses N-glycosidase activity against 28S rRNA of 60S ribosomes in the cytosol, resulting in the inhibition of protein synthesis in eukaryotic cells and in the activation of proinflammatory signaling cascade referred to as the ribotoxic stress response (RSR) [16]. The five B subunits (StxB) form a pentamer that binds to globotriaosyl ceramide receptors (Gb3) on the cell membrane. STEC expresses two types of Stx proteins (Stx1 and Stx2) and their variants, being Stx2 more virulent and epidemiologically more relevant than Stx1 [17]. STEC has been classified into pathotypes and clades according to Stx-phage type that determines Stx-variant and level of Stx-production, Stx2 overexpression is common to STEC strains from clade 8, highly associated with HUS.
Previous studies from Rivas M, et al. [18] have demonstrated that E. coli O157:H7 harboring the Stx2a/ Stx2c genes was prevalent among diarrhea and HUS pediatric patients in Argentina [18]. In the same way, Tarr, et al. in 936 E. coli O157: H7 patients found a considerable increase in HUS risk associated with the Stx2a genotype as compared to the Stx1a/Stx2a genotype-two of the most common genotypes in USA and Japan [19]. Recently, Pianciola, et al. Reported O157 strains isolated in Argentina and found the Stx2a/Stx2c genotype was prevalent in human and bovine strains, 87.6% of the strains belonged to the hypervirulent clade 8. These data may allow us to understand the causes of the epidemiological situation related to HUS in Argentina [20]. This STEC serotype is responsible for severe disease, rapid progression, and high hospitalization rates [3].
Shiga-Toxin Action
After ingestion of contaminated food with STEC, the bacteria lodge in the colon, where it adheres to the epithelium and multiplies. The incubation period between infection and diarrhea is 1 to 8 days, with intestinal shedding of the bacteria continuing for 3 or more weeks. The first step consists of the interaction and adherence of STEC to the intestinal epithelium, a process that is characterized by the production of lesions called “attaching and effacing” or A/E lesions.
After bacterial lysis, Shiga toxins are released into the intestinal lumen, and its B subunit binds to its STEC specific receptor globotriaosylceramide (Gb3), located primarily in lipid rafts at the plasma membrane and induced endocytosis. Stx could cross the intestinal epithelium by both transcellular and paracellular routes [21]. After Shiga toxins (Stxs), released from STEC, further damage the vascular network of the intestinal mucosa, causing hemorrhagic colitis.
Expression of Gb3 in humans is restricted to microvascular endothelial cells, podocytes, platelets, germinal center B lymphocytes erythrocytes (where it constitutes the rare Pk antigen), and neurons [22]. The reason behind the specific distribution of glycosphingolipid in human tissues are unknown. The estimated half-life of Stx in serum is less than 5 min, as it rapidly diffuses to affected tissue, as a consequence of this, by the time patients develop HUS, Stx has disappeared from the serum. In Gb3-positive cells, the Stx-Gb3 complex induces membrane invagination that facilitates endocytosis. Then, the Stx-Gb3 complex is sent from early endosomes to the endoplasmic reticulum through retrograde transport, making it possible for Stx Gb3 to escape lysosomal degradation [23].
![Figure 1: Intracellular trafficking and cytotoxicity of Shiga toxin. 1. Shiga toxins consist of a monomeric enzymatically active A subunit, non-covalently linked to a pentameric B subunit. The B subunit binds to the glycosphingolipid globotriaosylceramide (Gb3). 2. Shiga toxin and its receptor are internalized (endocytosis), and Shiga toxin is activated through cleavage of the A subunit into 2 fragments by the protease furin. 3. Shiga toxin avoids the lysosomal pathway and is directed toward to the endoplasmic reticulum (retrograde transport). 4. The A1 subunit translocates into the cytoplasm (anterograde transport) where it can exert its cytotoxic effects. 5. The processed A1 fragment cleaves one adenine residue, thus inhibiting protein synthesis and triggering the ribotoxic and endoplasmic reticulum stress responses. 6. In addition to its ribotoxic effect, Shiga toxin activates multiple stress signaling and apoptotic pathways, and is responsible for the production of inflammatory cytokines by target cells. Modified from Joseph A, et al. [23].](/fulltextimages/11638/fig_1.png)
Figure 1: Intracellular trafficking and cytotoxicity of Shiga toxin.
- Shiga toxins consist of a monomeric enzymatically active A subunit, non-covalently linked to a pentameric B subunit. The B subunit binds to the glycosphingolipid globotriaosylceramide (Gb3).
- Shiga toxin and its receptor are internalized (endocytosis), and Shiga toxin is activated through cleavage of the A subunit into 2 fragments by the protease furin.
- Shiga toxin avoids the lysosomal pathway and is directed toward to the endoplasmic reticulum (retrograde transport).
- The A1 subunit translocates into the cytoplasm (anterograde transport) where it can exert its cytotoxic effects.
- The processed A1 fragment cleaves one adenine residue, thus inhibiting protein synthesis and triggering the ribotoxic and endoplasmic reticulum stress responses.
- In addition to its ribotoxic effect, Shiga toxin activates multiple stress signaling and apoptotic pathways, and is responsible for the production of inflammatory cytokines by target cells.
Modified from Joseph A, et al. [23].
As described by Joseph A, et al. [23] the catalytic A subunit is cleaved by the protease furin into two fragments: A1 and A2. In the endoplasmic reticulum, the disulfide bound between the two fragments is reduced. The A1 fragment translocates into the cytoplasm (anterograde transport) where it cleaves one adenine residue from the 28S RNA of the 60S ribosomal subunit, thus inhibiting protein translation and triggering the ribotoxic and endoplasmic reticulum stress responses [23]. The mechanism allowing Shiga toxins to bypass late endosomes and lysosomes is only partially known. Activation of the ribotoxic stress response leads to signalling through MAPK cascades, which appears to be critical for activation of innate immunity and regulation of apoptosis. Along with its ribotoxic effect, Shiga toxin activates multiple stress signaling and apoptotic pathways in target cells [23]. The pathophysiology of Shiga toxin trafficking and intracellular action is summarized in Figure 1.
The affinity of the binding between Gb3Cer and Stxs in the microvascular endothelium is 100 times higher than that in granulocytes [24]. Stxs can also bind to monocytes, resulting in the release of cytokines that upregulate Gb3Cer expression in endothelial cells.
Inflammatory and Thrombotic Response in STx-HUS Patients
Endothelial injury has been recognized as a trigger event in the microangiopathic process. Host endothelial cell inflammatory response to E. coli Stx and/or LPS contributes to the ongoing vascular damage from the infection, which results in HUS. The inflammatory response of host endothelial cells is included in the development of vascular damage observed in the infection, resulting in HUS [25]. Accordingly, human endothelial cells stimulated with subinhibitory concentrations of Stx elicit few, but reproducible changes in gene expression of chemokines; these findings indicate the important role of Stx in inducing a multifaceted host inflammatory response [26]. LPS augments Stx toxicity [27].
Activation of a strong inflammatory response leading to cytokines and chemokines release, the leukocytes and monocytes recruitment, and the activation of complement and thrombotic cascades have been shown during the early stage of STEC-HUS [28]. Blood polymorphonuclear neutrophils (PMN) and monocytes/macrophages are sensitive to Stx and may be key players in HUS outcomes.
Increased peripheral polymorphonuclear leucocyte (PMN) count at onset in childhood HUS is one of the most consistent parameters of inflammation associated with a poor prognosis [29]. PMN may contribute to renal inflammation and endothelial injury during HUS, since their great cytotoxic potential through exocytosis of granules- containing proteases and other enzymes, as well as high release of ROS after activation. A study from Palermo, et al. revealed a marked deactivation of PMN in severe cases of HUS, suggesting that the study of the functional state of PMN could be of prognostic value. Previous data reporting decreased intracellular content of enzymes and antigens supports the concept that PMN count from Stx-HUS patients has been previously activated and then degranulated [30]. According to these results, it has been proposed that a strong initial activating stimulus would induce strong activation of PMN, which in turn would drive to a more severe clinical course. Previously in a clinical study increased TNF-α and IL-8 was shown in patients at HUS onset. The levels of the proinflammatory cytokines TNF-α and IL-8 decreased in these patients after 3 days, with no significant differences after 3 and 10 days from HUS onset, compared with healthy children [31].
HUS patients showed an elevation of CD14+ CD16+ monocytes, a subset considered to represent activated, more mature cells with characteristics that resemble macrophages and dendritic cells. Correlation between monocytosis and HUS severity has been shown in Stx-HUS patients [32], together with changes in the expression of chemokine receptors on these cells [32]. TNF-α and IL- 10 production by monocytes increases in parallel with the severity of disease in Stx-HUS children, in such a way that patients with moderate-to-severe disease have the greatest number of TNF-α- producing monocytes [33]. Recently we demonstrated increased generation of intracellular cytokines TNF-α and IL-6 in the CD14+ monocytes from Stx HUS patients during the early stage, day 1 to day 4. A significant reduction of intracellular monocyte cytokines after 10 days with intracellular expression of TNF-α levels in a time dependent manner from day 4 to day 10 [34].
Vascular damage triggered by Stx not only induces the production of cytokines and chemokines by endothelial cells but also promotes the release of thrombin and increases fibrin concentrations. The presence of high levels of plasminogen activator inhibitor-1 (PAI-1) blocks fibrinolysis and accelerates thrombosis. Stxs directly activate platelets [35]. Thromboresistance in endothelial cells induced by leukocyte- dependent inflammation leads to microvascular thrombosis, as reported on human microvascular endothelial cells pre-exposed to Stxs [3]. In addition, it has been recently shown that Stx can directly interact with the von Willebrand factor (VWF). This interaction causes a delay in the cleavage of VWF by a disintegrin and metalloproteinase, providing another explanation for thrombus formation in STEC-HUS [36].
A further potential mechanism involved in STEC-HUS thrombus formation is the release of neutrophil extracellular traps (NETs) by PMN, in a ROS/NADPH-dependent manner. NETs are a meshwork of DNA fibers comprising histones and granule proteins, formed by a cell-death program which proceeds from the dissolution of internal membranes followed by chromatin decondensation and cytolysis [37]. They have strong antimicrobial and/or immunomodulatory properties, although, high amounts of them seem to be associated with pathophysiological conditions. Inside the microvasculature, NETs act as a stimulus for thrombus formation. Reports of decreased degradation and increased formation of NETs have suggested a role for NETs in thrombotic microangiopathic disorders [38]. A study of STEC/ HUS patients found that 50% of them failed to degrade NETs, associated with decreased kidney function [39].
Innate Immunity in HUS
Toll-Like Receptor 4 (TLR4) Expression
Both bacterial lipopolysaccharide (LPS) and polymorphonuclear neutrophils play a key role in the full development of HUS. Therefore, synergism between LPS and Stx2 has been demonstrated as a consequence of the enhancement of Stx2 renal toxicity [40]. LPS is the major component of the outer membrane of gram-negative bacteria, being a critical actor in the pathogenesis of gram- negative sepsis [41]. The recognition of LPS by innate immune cells is vital for host defenses against gram-negative bacteria. On the surface of immune cells, pattern recognition receptors [toll- like receptors (TLRs)] sense the presence of these bacterial components and activate common pathways to mediate responses [41], with TLR4 an essential receptor for LPS signal transduction, LPS binding to TLR4 activates an intracellular signal, which travels from the cytoplasm to the nucleus, binds to initiation sites, and increases transcription of inflammatory cytokines. Clearly, neutrophil and monocyte cells express TLR4, and more importantly, these cells respond to LPS, providing insight into the complex pathology in sepsis and potentially in HUS [42]. Evidence has been reported that human platelets bind LPS from enterohemorrhagic EHEC through a complex of TLR4 and CD62, leading to their activation [43].
Recently, we investigated TLR4 surface receptor expression on peripheral blood neutrophils and their ability to modulate inflammatory cytokine release in pediatric patients 1,3 and 10 days after HUS onset The isolated leucocytes from the HUS-onset patients exhibited significantly higher mRNA TLR4 expression than the controls. Moreover, TLR4 protein expression on neutrophils determined by flow cytometry was upregulated, driving dependent proinflammatory cytokine, tumor necrosis factor-alpha, IL-8 increase, and decreased anti-inflammatory IL-10 release at HUS onset compared with patients with STEC E. coli (EHEC) diarrhea, and healthy children. Conversely, a significant reduction of the neutrophil TLR4 receptor expression and lack of cytokine-responsive element activation was shown in patients 3 and 10 days after the HUS onset. These results suggest that TLR4 expression may be differently regulated on neutrophils. They could be dynamically modulated across the early development of HUS on neutrophils, resulting in the negative regulation preceded by TLR4 overactivation [31].
The receptor-ligand intracellular membrane trafficking and routing is crucial to reach a controlled inflammatory response and regulated termination of inflammatory signaling. As described by Vasquez-Carvallo, et al. Modulation of TLR4 activation and their downstream-related signaling pathways include several mechanisms, such as soluble decoy receptors, transmembrane regulators, cellular trafficking, destabilization of adaptor proteins, ubiquitination, dephosphorylation, transcriptional regulation, and feedback inhibition [44].
The TLR4-LPS complex is rapidly internalized and TLR4- induced inflammatory signaling is stopped by targeting the complex for degradation. Endosomes play an essential role in this process, serving as sorting facilities for the biosynthetic and endocytic pathways. The LPS/TLR4 complex internalized by endocytosis in early endosomes is then delivered to late endosomal/lysosomal for degradation or toward the Golgi apparatus for recycling [45].
Rab7, a small GTPase localized to late endosomes, regulates the later stages of the endocytic pathway and it is involved in trafficking and lysosomal degradation of several kinds of receptors Rab7b may negatively regulate TLR4 signaling, which promotes TLR4 targeting to lysosomes for degradation [46].
We investigated the role of Rab7b in LPS-initiated TLR4 signaling in monocytes during the acute course of Shiga toxin-associated HUS. We found that at the very beginning of the disease, on days 1 and 4, Rab7b colocalizes with TLR4 in intracellular vesicles with maximal colocalization at day 4. These results suggest a localization of TLR4 within Rab7b- coated vesicles. Both proteins run together in intracellular trafficking directed towards degradation Likely the higher TLR4 observed at the surface level by flow cytometry could demonstrate the recycling of the receptor in a step before the proteolytic degradation; and/or, it may be possible that the sustained stimulus of the pathogen could generate de- novo synthesis, which also would contribute to the increased receptor expression in the cell membrane on day 4. As the disease progresses, surface TLR4 decreases as well as the monocyte intracellular cytokine inflammatory response. Monocyte cell analysis on day 10 showed surface TLR4 expression and intracellular proinflammatory cytokines expression near controls. Lower expression of Rab7b and TLR4 and minimal and punctual colocalization of both proteins were observed with no differences related to control. In this way, the early colocalization of Rab7b/TLR4 may account for an adequate/successful trafficking of TLR4 to the degradation pathway leading to the downregulation of proinflammatory state or inflammation during the early follow-up (10 days) of the disease (Figure 2).
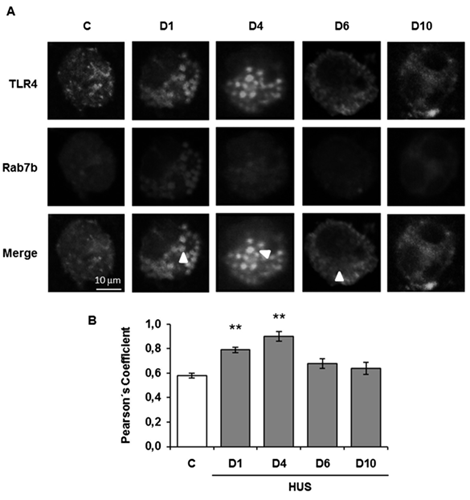
TLR4 and Rab7b colocalization in monocytes was determined throughout the progression of HUS and in controls by indirect immunofluorescence and confocal microscopy. Specific rabbit anti TLR4/anti rabbit FITC (green), mouse anti Rab7b/anti mouse Cy3 (red), and Hoechst to visualize the nuclei (blue) were used. A. Representative immunofluorescence of both proteins in monocytes from one HUS patient and one control. The arrows indicate the colocalization areas. Scale bar 10μm. Magnification 600X. B. Graphic bar shows the TLR4 and Rab7b colocalization, which was determined by using the Pearsons’ Coefficient. **p < 0.01 vs control. 20 cells from 3 independent cultures were analyzed for each day of the HUS follow-up. In addition, 20 cells from 5 independent cultures in the control group were evaluated. Data are expressed as the mean ± SEM.
Our findings suggest that Rab7b participates as a negative regulator in the control of the TLR4 endocytic pathway in pediatric HUS patient monocytes. The resulting decline in monocyte cell cytokine generation, is demonstrated by the induction of the TLR4 receptor endocytosis during the early follow-up of HUS [34].
Complement
Activation of a strong inflammatory response causing the release of cytokines and chemokines, the recruitment of leukocytes and monocytes, and the activation of complement and thrombotic cascades have been shown during the early stage of STEC-HUS [28]. There is growing evidence of alternative complement pathway activation in STEC-HUS. Plasma levels of Bb and C5b-9, two complement pathway products, and C3-bearing microparticles from platelets and monocytes were found to be elevated in patients suffering from STEC-HUS. Both decreased at recovery but were not associated with disease severity. Children with STEC-HUS and reduced levels of C3 are at risk for neurological involvement and severe clinical manifestations [47].
Recently serological profiles (C3, FH, FI, AP activity, C3d, C3bBbP, C3b/c, αFH) and genetic profiles (CFH, CFI, CD46, CFB, C3) of the alternative complement pathway were prospectively determined in the acute and convalescent phase of disease in children newly diagnosed with STEC- HUS. In 28 % of STEC-HUS patients, it was identified a genetic and/or acquired complement abnormality, the levels of investigated alternative pathway (AP) activation markers were elevated in the acute phase and normalized in remission [48].
Although the pathogenic role of complement activation is still poorly understood, new data from the most recent outbreaks have suggested a relationship between complement activation and CNS compromise.
STEC-HUS: Clinical Features and Diagnosis
Patients exposed to STEC who will develop colitis after a median incubation of 4 d (1 to 10 d) usually present with painful diarrhea and abdominal cramping, vomiting and fever are less frequent, the disease is usually limited to the colon. Bloody diarrhea is not a defining feature of STEC-HUS, in 20%–30% of patients it may never occur. The percentage of patients that progress from STEC-diarrhea to STE- HUS ranged from 3 to 9 % in a series of sporadic cases to about 20 % or more in some outbreaks [49]. Microbiology Bacteriological investigation is the gold standard for the diagnosis of STEC infections. A prompt and accurate etiological diagnosis is needed in the face of a thrombotic microangiopathy syndrome in order to fit the initial treatment that is specific to each etiology. The rapid identification of EHEC allows for isolation measures that will prevent the further spreading of the pathogen, as well as avoidance of antibiotic therapy and antimotility agents in cases of STEC- related disease.
Fecal culture usually needs to be combined with a polymerase chain reaction (PCR) to detect the Stx-encoding gene, improve the detection rate, and further distinguish non-O157 from O157 infections [11]. As in Europe and the United States, in Argentina O157:H7 is a dominant STEC serotype that causes D+HUS [11].
Diagnosis relies on the detection and distinction of genes encoding Shiga toxins (stx1 and/or stx2) by polymerase chain reaction (PCR) following stool enrichment. The results are then available within 12 to 24 hours. Taking into account the primer used, PCR can also detect stx subtypes; virulence-associated genes such as eae encoding for intimin, ehxA encoding enterohemolysin, and aggR encoding for aggregative adherence fimbria I; or the specific O group of the pathogen [11]. All these features may detect risk factors for HUS evolution. From the first day of diarrhea to the development of HUS, the average time is 5–13 days nevertheless, STEC and its toxins in the intestine decrease rapidly within a week after the onset of symptoms [50].
Detection of antibodies directed against LPS (O-groups) is of great diagnostic value: IgM appears soon after the infection and peak at day 9, whereas IgG appears from day 8 and persist several weeks after infection [11, 50]. After two to three weeks, repeated serology may demonstrate an increase in antibody titers. The combination of serology with standard fecal diagnostic tests could be specifically useful when patients present late in the course of the disease and at the time of HUS.
STEC-HUS Diagnosis
In hemolytic uremic syndrome (HUS), characterized by thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury (AKI) the diagnosis is usually made 6 to 10 days after the onset of diarrhea. The laboratory criteria for HUS diagnosis included acute onset, hemoglobin less than 10 g/dL, characterized by red blood cell fragmentation with schistocytes (burr or helmet cells) in the peripheral blood smear, signs of increased hemolysis with LDH > 600 UI/L. Other indicators of intravascular hemolysis include hyperbilirubinemia, reticulocyte counts uniformly elevated, and low or undetectable haptoglobin concentrations. To confirm the microangiopathic nature of the hemolytic anemia, the detection of fragmented erythrocytes together with a negative Coombs’ test is crucial. However, patients with pneumococcal-HUS may have a positive direct Coombs’ test result. Moderate leukocytosis may accompany hemolytic anemia. Consumption of platelets in thrombi causes thrombocytopenia with platelet count less than 150 × 109/L [3, 10].
Renal Involvement ranges from proteinuria, leukocyturia, and microscopic hematuria to oliguria and acute kidney injury. Acute kidney injury (AKI) in STEC-HUS patients requires renal replacement therapy (RRT) during the acute stage, with a mean duration of oliguria or RRT of 9–10 days. A 15% percent of children suffer from high blood pressure [51].
Chronic kidney disease can become apparent at different intervals after the acute HUS episode. It is related to the duration of anuria, receipt of kidney-replacement therapy, or both during HUS [51]. A recent report suggests that chronic kidney injury can be detected in up to one-third of children who have had HUS but did not receive kidney replacement therapy [52].
Hypertension, proteinuria, and a reduced glomerular filtration rate can be manifested years after the acute episode of STEC-HUS. Hence, it is mandatory to follow up patients throughout their childhood, for renal sequelae.
The kidney and the brain are the organs most vulnerable to STEC-HUS but several other organ involvements have been described and need to be considered when evaluating patients with STEC- HUS. This disease is a systemic disorder in which severe forms can be associated with multiorgan involvement during the acute stage of the disease [3].
Neurologic involvement is one of the most terrible complications of STEC-HUS. It is a main contributor to the morbidity and mortality of the disease. Central nervous system disturbances are usually present early in the course of the illness; including stroke, seizure, and coma. Other characteristics include pyramidal and extrapyramidal features, diplopia, dysphasia, and facial nerve palsy. Severe neurological involvement, mainly in the form of persistent encephalopathy, brain hemorrhage or infarction, and anoxic brain injury, is associated with worse disease severity and higher mortality [53]. Brain MRI typically reveals bilateral hypersignal on T2-weighted and hypo signal on T1-weighted images of basal ganglia, thalami, and brainstem sometimes extending to the surrounding white matter. Additionally, MRI may display images of high blood pressure complications such as reversible posterior leukoencephalopathy syndrome and cerebral hemorrhage. Fatal outcome is recorded in around 20% of patients with neurologic involvement, and severe sequelae are observed in about 27% of these patients. During the O104:H4 outbreak, in which half of the patients developed neurologic symptoms, epileptic seizures were seen in 20%, and cognitive impairment or aphasia in 67.3% [54]. From 362 STEC-HUS patients treated at Mendoza, Argentina forty-one patients developed neurologic symptoms. The earlier clinical findings were related to cognitive dysfunction, such as disorientation, restlessness, lethargy, and drowsiness. Thirty-three patients suffered from generalized tonic-clonic seizures and four patients, focused tonic-clonic seizures Hemiparesis was present in one patient. One patient suffered from extrapyramidal syndrome. Twelve patients died, three patients had severe sequelae [3]. Neurologic complications often parallel renal failure and are exceedingly rare in the absence of AKI. Neurologic symptoms as the unique manifestation of thrombotic microangiopathy should prompt clinicians to consider the diagnosis of thrombotic thrombocytopenic purpura (TTP) rather than STEC-HUS.
STEC-HUS patients suffer from high blood pressure during the early stage of the disease, and it is due to different conditions including volume overload and electrolyte imbalance in the setting of underlying TMA involving the kidneys. The incidence of hypertension in STEC-HUS has been reported early after ICU admission.
In our group of patients, 41% developed high blood pressure above the 95th percentile, one patient also suffered from dilated cardiomyopathy during his ICU stay.
Cardiac Involvement usually appears a week or more after the onset of HUS. Clinical manifestations (cardiac ischemia, rhythm disorders, cardiac arrest) seem to occur in a low group of STEC-HUS pediatric patients, less than 10%. Pericardial compromise with cardiac tamponade has also been recorded. Cardiomyopathy and myocarditis are described and may occur several weeks after onset. They may or may not be secondary to hypertension or vascular volume overload Although rarer, acute myocardial infarction is another potentially life-threatening complication of STEC- HUS.
Pulmonary complications include respiratory distress syndrome, central volume overload, pleural effusion, and pulmonary hemorrhage the last one, cause of death.
Digestive tract complications: The whole digestive tract can also be involved. Severe hemorrhagic colitis, bowel necrosis and perforation, rectal prolapse, peritonitis, and intussusception are included as serious digestive tract complications [55]. Rarely, catastrophic intestinal events such as bowel necrosis and perforation occur. In our series, bloody diarrhea was present in the prodromal phase of the 44 STEC-HUS patients. Of the nine patients who showed digestive involvement, 6 patients underwent enterorrhagia and hemorraghic colitis, 1 patient had rectal prolapse, 1 patient pancreatitis, and the last patient severe hepatic failure.
Biological pancreatitis, as well as elevated liver enzymes, occur in a low percent of STEC-HUS patients, do not commonly result in organ failure. Survivors of STEC-HUS (but not uncomplicated EHEC infection) have a significantly increased incidence of diabetes possibly as a consequence of thrombosis of vessels supplying the islets of Langerhans as evidenced in autopsy series. Histopathology Renal biopsy is only performed in the context of STEC- HUS in the case of diagnostic uncertainty, which makes its histopathological description rare and potentially biased. Nevertheless, STEC-HUS patients seem to display unspecific features of thrombotic microangiopathy, such as glomerular capillary thromboses with a widened subendothelial space, endothelial swelling, and congested glomeruli. Necrosis of capillary walls, with luminal narrowing and thrombosis, is also characteristic. Cortical infarcts can be seen in severe and fatal cases.
Supportive Therapy
Once the disease is established, supportive therapy is the mainstay of the treatment of STEC-HUS patients. It is mostly responsible for the improved prognosis in recent years. Hospitalization is mandatory, in specialized centers, and intensive care is often required. Treatment relies principally on supportive care, which includes fluid resuscitation, the correction of electrolyte abnormalities, and the control of hypertension. Blood or platelet transfusions and renal replacement therapy (RRT) are often required.
Administration of intravenous fluid and sodium as soon as a STEC infection is suspected (that is within the first 4 days of illness, even before culture results are available) seems to limit the severity of ARF and the need for renal replacement therapy. Intravenous fluid expansion up to, and including, the day of STEC-HUS diagnosis has also proven to lessen the need for renal replacement therapy (RRT) and reduce central nervous system-associated complications, as dehydration has been associated with mortality. Other authors reported that increased volume expansion with intravenous isotonic saline during the diarrheal phase attenuates but does not prevent renal injury. Avoidance of hypotonic fluids, due to the increased risk of hyponatremia in hospitalized children, is becoming standard in pediatric care.
Early use of isotonic fluids could be recommended in patients with dysentery, starting from the onset of bloody diarrhea to the day of onset of HUS, but when AKI is established, it should be balanced against the risk of fluid overload.
Packed red-cell transfusions are administered to most patients with STEC-associated HUS Importantly, restrictive thresholds of 7 g/dL, advocated in the recent American Association of Blood Banks (AABB) guidelines. Platelet infusions should be limited to patients with hemodynamically significant bleeding because most HUS complications are related to thrombotic injury, platelet transfusion may exacerbate the disease. Antimicrobial agents in the setting of HUS have sparked an ongoing controversy. Multiple studies have shown an association between antibiotic administration and an increased risk of HUS among patients infected with high-risk STEC. The use of antibiotics may lead to an increase in Stx release from dead bacteria or to alterations in the intestinal flora that are conducive to the further attachment of STEC to the intestinal wall, the induction of phage production, and the expression of Stx genes, which may lead to disease progression and deterioration. Clinical studies that segregated the role of specific antibiotic classes have demonstrated that ß-lactams, metronidazole, and trimethoprim-sulfamethoxazole were associated with the most significant risk for HUS, whereas azithromycin and aminoglycosides were protective against HUS development [56].
In a recent review of antibiotics and HUS, Tarr and Freeman based on evidence from multiple retrospective cohort studies recommended against the use of antibiotics in patients with a confirmed or suspected STEC infection. The reason is that there is still no data that convincingly shows that antibiotics are superior to no antibiotic treatment at all, and many studies have shown that antibiotics increase the risk of developing HUS [57].
The use of antimotility agents has been associated with an excess risk of HUS development in children infected with EHEC. In accordance with the 2014 recommendations of the European Society for Pediatric Gastroenterology for acute gastroenteritis should therefore be discouraged. Nonsteroidal anti-inflammatory drugs can cause acute kidney injury during gastrointestinal infection and are best avoided.
Blood Pressure Control Hypertension is a well- established contributor to thrombotic microangiopathy lesions and could also partly account for the occurrence of posterior reversible encephalopathy syndrome (PRES). It should, therefore, be managed with appropriate medication in the acute stage, such as calcium receptor blockers or diuretics in the case of fluid overload.
Renal Replacement Therapy
Around 40~71% of STEC-HUS patients require RRT. When patients develop oliguria AKI, fluid overload, refractory hyperkalemia, or uremia, RRT is required. It includes peritoneal dialysis (PD), hemodialysis (HD), and continuous renal replacement therapy (CRRT). Ongoing evidence indicates that there is no significant difference in mortality for PD, HD, and CRRT in AKI. In Argentina, the country with the highest incidence of STEC-HUS in the world, PD is the most commonly used method and has always been the main RRT method for the pediatric treatment of AKI.
Hemodialysis should be considered when rapid fluid and solute removal is required and has been more commonly used in older children and adults. However, thrombocytopenia- associated bleeding during catheter insertion, catheter malfunction, and catheter-related sepsis are potential complications.
CRRT is the most appropriate treatment for critically ill patients with multiorgan dysfunction and hemodynamic instability. Its significant advantages are better hemodynamic stability and reduced cross-cell solute migration, avoiding the increased intracranial pressure that may be caused by HD. CRRT provides more efficient solute removal, liquid ultrafiltration, and easier fluid balance control than PD. Chronic kidney disease can become apparent at variable intervals after the acute HUS episode.
Specific Therapies
Despite major achievements in the understanding of the pathophysiology of the disease, the quest for a specific treatment remains difficult to find.
Plasma Exchange and Immunoadsorption
Early institution of therapeutic plasma exchange (TPE) could theoretically aid in the disease- modifying effects related to the removal of proinflammatory cytokines, and prothrombotic factors thereby limiting the inflammatory response [58]. Even though plasma exchange is beneficial in patients with thrombotic thrombocytopenia purpura because it corrects the absence or dysfunction of ADAMTS13, evidence to support its use in those with STEC-associated HUS is extremely limited.
Therapeutic plasma exchange has been applied most commonly as a salvage therapy for pediatric patients who develop neurological manifestations. In our study, TPE was undertaken in 37 patients with severe neurological compromise from the very beginning after admission to ICU. Of the 37 TPE-treated patients, 11 were treated within 2 hours after the first neurological sign; two died, three survived with severe sequelae, and seven with moderate disabilities.
Currently, no definitive answers concerning its efficacy can be given, highlighting the need for well-performed randomized controlled trials.
Complement Blockade Therapy
Eculizumab an anti-C5 monoclonal antibody, is highly effective in atypical HUS, but only rarely do patients with STEC-associated HUS have complement gene variants that are pathogenic. The O104:H4 outbreak provided an unprecedented basis for clinical investigation, and many patients were treated with complement blockade therapy. If the analysis of the German registry [59] did not support the use of eculizumab in adult STEC-HUS cases, early treatment was associated with rapid and efficient recovery in French patients [60] and children with central nervous system involvement. A concern with eculizumab treatment is the risk of infection with encapsulated bacterial organisms, particularly Neisseria meningitis, as a result of terminal complement blockade Therefore, patients must receive meningococcal vaccination before being treated with Eculizumab at least 1 week before treatment. Considering that the majority of patients recover with supportive treatment, the risks and benefits of Eculizumab need to be fully evaluated before its use, especially for those with complement activation, neurological involvement, and a high risk of death.
In the meantime, randomized controlled trials of Eculizumab after the stratification of disease severity will provide more convincing evidence, and positive results are expected in some ongoing clinical trials.
Shiga Toxin Neutralization
Antibodies can neutralize Stxs in the serum and potentially even in the gut, making these molecules powerful weapons against toxins. Strategies for neutralizing extraintestinal toxin early in the course of STEC infection are attractive since systemic toxemia is likely to precede HUS. Only a minority of infected children have quantifiable levels of circulating Shiga toxin 2 in the first days of diarrhea, and even among these children, toxemia is short-lived. Thus, the opportunity to neutralize extraintestinal Shiga toxin might be limited.
Randomized controlled trials are needed to confirm the potential role of Stx-neutralizing (monoclonal) antibodies (STmAb) in STEC-HUS patients.
Conclusions
Shiga toxin-associated HUS remains a global health concern. Diagnostic approaches include the testing of all children with bloody diarrhea for bacterial pathogens with the use of techniques that can identify O157 and non-O157 STEC, and toxin genotypes reports when STEC are identified, constitute important components of care.
Initial recognition of severe forms of STEC-HUS associated with multiorgan involvement during the acute phase of the disease is mandatory. Although extensive improvements in the understanding of the pathophysiology and the encouraging results in preclinical models and ongoing clinical trials, a specific treatment is still absent.
Further studies will require new therapeutic alternatives to improve mortality and avoid sequelae in critically ill pediatric patients. Controlled studies need to be continued.
References
-
Ruggenenti P, Noris M, Remuzzi G (2001) Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int 60(3): 831-846.
-
Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, et al. (2016) An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatric Nephrology 31(1): 15-39.
-
Luna M, Kamariski M, Principi I, Bocanegra V, Vallés PG, et al. (2021) Severely ill pediatric patients with Shiga toxin-associated hemolytic uremic syndrome (STEC- HUS) who suffered from multiple organ involvement in the early stage. Pediatr Nephrol 36(6): 1499-1509.
-
Oualha M, Pierrepont S, Krug P, Gitiaux C, Hubert P, et al. (2018) Postdiarrheal hemolytic and uremic syndrome with severe multiorgan involvement and associated early risk factors. Archives de Pediatrie 25(2): 118-125.
-
Scully M, Cataland S, Coppo P, Rubia JDL, Friedman KD, et al. (2017) Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost 15(2): 312-322.
-
Copelovitch L, Kaplan BS (2010) Streptococcus pneumoniae-Associated hemolytic uremic syndrome: Classification and the emergence of serotype 19A. Pediatrics 125(1): 174-182.
-
Noris MRG (2009) Atipycal hemolytic-uremic syndrome. N Engl J Med 361(17): 1676-1687.
-
Michael M, Bagga A, Sartain SE, Smith RJH (2022) Haemolytic uraemic syndrome. The Lancet 400(10364): 1722-1740.
-
Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, et al. (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5(10): 1844-1859.
-
Mele C, Remuzzi G, Noris M (2014) Hemolytic uremic syndrome. Semin Immunopathol 36(4): 399-420.
-
Rivas M, Pichel M, Colonna M, Casanello AL, Alconcher LF, et al. (2023) Surveillance of Shiga toxin-producing Escherichia coli associated bloody diarrhea in Argentina. Rev Argent Microbiol 7541(23): 00028-00037.
-
Palermo MS, Exeni RA, Fernández GC (2009) Hemolytic uremic syndrome: Pathogenesis and update of interventions. Expert Rev Anti Infect Ther 7(6): 697-707.
-
Rivas M, Sosa-Estani S, Rangel J, Caletti MG, Vallés P, et al. (2008) Risk factors for sporadic Shiga toxin-producing Escherichia coli infections in children, Argentina. Emerg Infect Dis 14(5): 763-771.
-
Karch H, Denamur E, Dobrindt U, Finlay BB, Hengge R, et al. (2012) The enemy within us: Lessons from the 2011 European Escherichia coli O104:H4 outbreak. EMBO Mol Med 4(9): 841-848.
-
Scheiring J, Andreoli SP, Zimmerhackl LB (2008) Treatment and outcome of Shiga-toxin-associated hemolytic uremic syndrome (HUS). Pediatric Nephrology 23(10): 1749-1760.
-
Smith WE, Kane AV, Campbell ST, Acheson DWK, Cochran BH, et al. (2003) Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect Immun 71(3): 1497-1504.
-
Ibarra C, Amaral MM, Palermo MS (2013) Advances in pathogenesis and therapy of hemolytic uremic syndrome caused by Shiga toxin-2. IUBMB Life 65(10): 827-835.
-
Rivas M, Miliwebsky E, Chinen I, Roldán CD, Balbi L, et al. (2006) Characterization and epidemiologic subtyping of Shiga toxin-producing Escherichia coli strains isolated from hemolytic uremic syndrome and diarrhea cases in Argentina. Foodborne Pathog Dis 3(1): 88-96.
-
Tarr GAM, Stokowski T, Shringi S, Tarr PI, Freedman SB, et al. (2019) Contribution and interaction of shiga toxin genes to Escherichia coli O157:H7 Virulence. Toxins Basel 11(10): 607.
-
Pianciola L, Astek BAD, Mazzeo M, Chinen I, Masana M, et al. (2016) Genetic features of human and bovine Escherichia coli O157: H7 strains isolated in Argentina. International Journal of Medical Microbiology 306(2): 123-130.
-
Lee MS, Tesh VL (2019) Roles of shiga toxins in immunopathology. Toxins Basel 11(4): 212.
-
Obata F, Tohyama K, Bonev AD, Kolling GL, Keepers TR, et al. (2008) Shiga toxin 2 affects the central nervous system through receptor globotriaosylceramide localized to neurons. Journal of Infectious Diseases 198(9): 1398- 1406.
-
Joseph A, Cointe A, Kurkdjian PM, Rafat C, Hertig A, et al. (2020) Shiga Toxin-Associated Hemolytic Uremic Syndrome: A Narrative Review. Toxins Basel 12(2): 67.
-
Loo DMT, Monnens LA, Velden TJ, Vermeer MA, Preyers F, et al. (2000) Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 95(11): 3396-3402.
-
Zoja C, Buelli S, Morigi M (2010) Shiga toxin-associated hemolytic uremic syndrome: pathophysiology of endothelial dysfunction. Pediatr Nephrol 25(11): 2231- 2240.
-
Zoja C, Angioletti S, Donadelli R, Zanchi C, Tomasoni S, et al. (2002) Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via NF- kappaB dependent up-regulation of IL-8 and MCP-1. Kidney Int 62(3): 846- 856.
-
Clayt F, Pysher TJ, Lou R, Kohan DE, Denkers ND, et al. (2005) Lipopolysaccharide upregulates renal shiga toxin receptors in a primate model of hemolytic uremic syndrome. Am J Nephrol 25(6): 536-540.
-
Exeni RA, Fernandez-Brando RJ, Santiago AP, Fiorentino GA, Exeni AM, et al. (2018) Pathogenic role of inflammatory response during Shiga toxin-associated hemolytic uremic syndrome (HUS). Pediatr Nephrol 33: 2057-2071.
-
Buteau C, Proulx F, Chaibou M, Raymond D, Clermont MJ, et al. (2000) Leukocytosis in children with Escherichia coli O157:H7 enteritis developing the hemolytic-uremic syndrome. Pediatric Infectious Disease Journal 19(7): 642-647.
-
Fernández GC, Gómez SA, Rubel CJ, Bentancor LV, Barrionuevo P, et al. (2005) Impaired neutrophils in children with the typical form of hemolytic uremic syndrome. Pediatric Nephrology 20(9): 1306-1314.
-
Vallés PG, Melechuck S, González A, Manucha W, Bocanegra V, et al. (2012) Toll-like receptor 4 expression on circulating leucocytes in hemolytic uremic syndrome. Pediatr Nephrol 27(3): 407-415.
-
Fernández GC, Ramos MV, Gómez SA, Dran GI, Exeni R, et al. (2005) Differential expression of function-related antigens on blood monocytes in children with hemolytic uremic syndrome. J Leukoc Biol 78(4): 853-861.
-
Fernández GC, Ramos MV, Landoni VI, Bentancor LV, Fernández-Brando RJ, et al. (2012) Cytokine production is altered in monocytes from children with hemolytic uremic syndrome. J Clin Immunol 32(3): 622-631.
-
Manzano AFL, Lorenzo AFG, Bocanegra V, Costantino VV, Cacciamani V, et al. (2019) Rab7b participation on the TLR4 (Toll-like receptor) endocytic pathway in Shiga toxin-associated Hemolytic Uremic Syndrome (HUS). Cytokine 121: 154732.
-
Karpman D, Papadopoulou D, Nilsson K, Sjögren AC, Mikaelsson SCL, et al. (2001) Platelet activation by Shiga toxin and circulatory factors as a pathogenetic mechanism in the hemolytic uremic syndrome. Blood 97(10): 3100-3108.
-
Lo NC, Turner NA, Cruz MA, Moake J (2013) Interaction of shiga toxin with the a-domains and multimers of von Willebrand Factor. Journal of Biological Chemistry 288(46): 33118-33123.
-
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, et al. (2007) Novel cell death program leads to neutrophil extracellular traps. Journal of Cell Biology 176(2): 231- 241.
-
Ramos MV, Mejias MP, Sabbione F, Fernandez-Brando RJ, Santiago AP, et al. (2016) Induction of Neutrophil Extracellular Traps in Shiga Toxin-Associated Hemolytic Uremic Syndrome. J Innate Immun 8(4): 400-411.
-
Leffler J, Prohászka Z, Mikes B, Sinkovits G, Ciacma K, et al. (2017) Decreased neutrophil extracellular trap degradation in shiga toxin-associated haemolytic uraemic syndrome. J Innate Immun 9(1): 12-21.
-
Landoni VI, Campos-Nebel M, Schierloh P, Calatayud C, Fernandez GC, et al. (2010) Shiga toxin 1-induced inflammatory response in lipopolysaccharide- sensitized astrocytes is mediated by endogenous tumor necrosis factor alpha. Infect Immun 78(3): 1193-1201.
-
Beutler B (2004) Innate immunity: An overview. Mol Immunol 40(12): 845-859.
-
Jerala R (2007) Structural biology of the LPS recognition. International Journal of Medical Microbiology 297(5): 353-363.
-
Ståhl AL, Svensson M, Mörgelin M, Svanborg C, Tarr PI, et al. (2006) Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 108(1): 167- 176.
-
Vázquez-Carballo C, Guerrero-Hue M, García-Caballero C, Rayego-Mateos S, Opazo-Ríos L, et al. (2021) Toll-Like Receptors in Acute Kidney Injury. Int J Mol Sci 22(2): 816.
-
Lu YC, Yeh WC, Ohashi PS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42(2): 145-151.
-
Bucci C, Bakke O, Progida C (2010) Rab7b and receptors trafficking. Commun Integr Biol 3(5): 401-404.
-
Netti GS, Santangelo L, Paulucci L, Piscopo G, Torres DD, et al. (2020) Low C3 Serum Levels Predict Severe Forms of STEC-HUS With Neurologic Involvement. Front Med Lausanne 7: 357.
-
Westra D, Volokhina EB, Molen RG, Velden TJAM, Jeronimus-Klaasen A, et al. (2017) Serological and genetic complement alterations in infection-induced and complement-mediated hemolytic uremic syndrome. Pediatric Nephrology 32(2): 297-309.
-
Mead PS GP (1998) Escherichia coli O157:H7. Lancet 10: 1207-1212.
-
Gould LH, Bopp C, Strockbine N, Atkinson R, Baselski V, et al. (2012) Update: Recommendations for Diagnosis of Shiga Toxin-Producing Escherichia coli Infections by Clinical Laboratories. MMWR Recomm Rep 58(12): 1-14.
-
Oakes RS, Kirkhamm JK, Nelson RD, Siegler RL (2008) Duration of oliguria and anuria as predictors of chronic renal-related sequelae in post-diarrheal hemolytic uremic syndrome. Pediatric Nephrology 23(8): 1303- 1308.
-
Alconcher LF, Lucarelli LI, Bronfen S (2023) Long-term kidney outcomes in non-dialyzed children with Shiga- toxin Escherichia coli associated hemolytic uremic syndrome. Pediatric Nephrology 38(7): 2131- 2136.
-
Khalid M, Andreoli S (2019) Extrarenal manifestations of the hemolytic uremic syndrome associated with Shiga toxin-producing Escherichia coli (STEC HUS). Pediatric Nephrology 34(12): 2495-2507.
-
Magnus T, Röther J, Simova O, Meier-Cillien M, Repenthin J, et al. (2012) The neurological syndrome in adults during the 2011 northern German E. coli serotype O104:H4 outbreak. Brain 135(6): 1850-1859.
-
Roessingh ABS, Lagausie P, Baudoin V, Loirat C, Aigrain Y, et al. (2007) Gastrointestinal complications of post- diarrheal hemolytic uremic syndrome. European Journal of Pediatric Surgery 17(5): 328-334.
-
Smith KE, Wilker PR, Reiter PL, Hedican EB, Bender JB, et al. (2012) Antibiotic treatment of escherichia coli O157 infection and the risk of hemolytic uremic syndrome, Minnesota. Pediatric Infectious Disease Journal 31(1): 37-41.
-
Tarr PI, Freedman SB (2022) Why antibiotics should not be used to treat shiga toxin-producing escherichia coli infections. Curr Opin Gastroenterol 38(1): 30-38.
-
Keir LS, Marks SD, Kim JJ (2012) Shiga toxin-associated hemolytic uremic syndrome: Current molecular mechanisms and future therapies. Drug Des Devel Ther 6: 195-208.
-
Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, et al. (2012) Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin- producing E. coli O104:H4 induced haemolytic-uraemic syndrome: An analysis of the German STEC-HUS registry. Nephrology Dialysis Transplantation 27(10): 3807- 3815.
-
Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, et al. (2014) Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: Outcome with eculizumab. Nephrology Dialysis Transplantation 29(3): 565-572.
- Results of 6-Month Follow-Up of Patients After B-Turp and Thulep
- The Effect of Drinking Water with a High Content of Antimony and Arsenic on the Dynamics of their Distribution in the Kidneys and the Renal Excretory Function in Rats
- Effectiveness and Safety of Tansurethral Thulium Laser Enucleation of the Prostate in the Treatment of BPH: Review
- A Systematic Review on Molecular Pathophysiology Involved in Chronic Kidney Disease and the Role of Animal Models in Drug Discovery to Manage in Chronic Kidney Disease - An Update
- Functional Development of Kidneys in Human Ontogenesis
- Testicular Metastasis: Uncommon Prostate Cancer Case Report