EPR-Proton Qubit's Role in Evolution and Age-Related Disease
Earth's surface acquired necessary life-giving volatile elements carbon, nitrogen, and sulfur from a collision with a Mercury-like planetary embryo ~ 4.4 billion y ago. Icy comets containing hydrocarbons collided with a cooling prebiotic Earth to create impact reactive environments that via classical anthropic causality introduced primordial ribozyme-like RNA complexes which could duplicate a few molecular units/24 hrs. Random classical processes introduced energetically accessible duplex RNA segments containing ketoamino―(entanglement) → enol−imine hydrogen bonds, where hydrogen bonded amino protons encountered quantum uncertainty limits.
Introduction
Quantum theoretical predictions have never been wrong when challenged by appropriately designed experimental tests [1] on reasonably well-isolated systems, e.g., single electrons, atoms, small molecules and near-perfect crystals [2]. Although quantum theory provides the most accurate description available for microscopic physical chemical reactive processes [3], its role in operational biological systems was previously considered to be negligible [4], since observable reactive biological systems are generally assumed to be embedded in “wet and warm” in vivo environments [5, 6]. Under these conditions, interactions between superpositions of entangled states [7, 8, 9, 10, 11] and water, ions and/or random temperature fluctuations [12] would cause rapid decoherence [13, 14], which would disallow quantum contributions to normally observed biological reactions. Nevertheless, operational molecular genetic systems exist [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26] that routinely exhibit “normal” molecular genetic reactions which are incompatible with classical Watson- Crick [27, 28], but are internally consistent with EPR- generated [29, 30, 31, 32, 33, 34] entangled proton qubits [35, 36, 37, 38, 39], subjected to measurements by Grover’s-type [40] quantum processors [41, 42, 43, 44, 45]. Dynamic quantum information processing [15, 16, 17] examples exhibited by ancient [46] T4 phage DNA [47, 48, 49, 50, 51] are also exhibited by (i) evolving rat – human microsatellite (short tandem repeats, STRs) distributions [38, 52], (ii) inherited Huntington’s disease [37, 53, 54] (CAG)n (n ≥ 36) repeats, and (iii) manifestation of distinguishable, entanglement- enabled “driver mutation”, versus classically-originated “passenger mutation” [26, 50, 55], exhibited by age-related cancers [35, 39, 56]. Based on the present and previousassessments [35, 36, 37, 38, 39], quantum information processing [40, 41, 42, 43, 44, 45] exhibited by prokaryotic [15, 16, 17, 20, 21] and eukaryotic [47, 48, 49, 50, 51, 52, 53, 54] genomic systems can be evolutionarily explained in terms of EPR-generated [29, 30, 31] entangled proton qubits originating in primordial RNA – ribozyme duplex segments [35, 36, 57, 58, 59, 60, 61, 62, 63], which subsequently were quantum mechanically processed by Grover’s-type [40] quantum readers. The molecular genetics history of observing [18, 19, 20, 21, 22, 23, 24, 25], but misidentifying, time-dependent EPR-generated [29, 30, 31]― keto-amino ― (entanglement) → enol-imine ― entangled proton qubits (Figure 1-4) has enabled perpetration of a falsifiable molecular genetics model, i.e., the classical molecular clock [15, 16, 17, 27, 28]. In these cases [18, 19, 20, 21, 22, 35, 36, 37, 38, 39, 49, 50, 51, 54], random classical processes [12] subjected metabolically inert [64], metastable hydrogen bonded amino (−NH2) DNA proton systems [65] to quantum uncertainty limits [2, 66], Δx Δpx ≥ ħ/2. This introduced a probability of direct quantum mechanical proton – proton interaction, yielding EPR- arrangements [29, 30, 31], keto-amino ― (entanglement) → enol−imine, observed [15, 16, 17] as G-C → G´-C´, G-C → *G-*C and A-T → *A-*T. (G´-C´, *G-*C,*A-*T― denote necessity of Hilbert space to describe dynamics of embedded entangled proton qubits; see Figure 2-4 for notation.)
![Figure 1: Schematic of “metastable” keto-amino and “ground state” enol-imine hydrogen bonds. (Amino protons encounter quantum uncertainty limits [2], ΔxΔpx ≥ ħ/2, introducing probabilities of EPR [29- 31,35-39] arrangements, _keto-amino ―(entanglement)→_ _enol−imine._ The asymmetric double-well potential represents an energy surface “seen by” a metastable hydrogen bonding amino proton, and a “ground state”, entangled enol or imine proton. Product enol and imine protons are entangled [7-10], and are each shared between two indistinguishable sets of electron lone-pairs, and thus, participate in entangled quantum oscillations, │+>⇄ │─>, between near symmetric energy wells, occupying intramolecular decoherence-free subspaces [67-69]).](/fulltextimages/1462/fig_1.jpeg)
Figure 1: Schematic of “metastable” keto-amino and “ground state” enol-imine hydrogen bonds. (Amino protons encounter quantum uncertainty limits [2], ΔxΔpx ≥ ħ/2, introducing probabilities of EPR [29, 30, 31, 35, 36, 37, 38, 39] arrangements, keto-amino ―(entanglement)→ enol−imine. The asymmetric double-well potential represents an energy surface “seen by” a metastable hydrogen bonding amino proton, and a “ground state”, entangled enol or imine proton. Product enol and imine protons are entangled [7, 8, 9, 10], and are each shared between two indistinguishable sets of electron lone-pairs, and thus, participate in entangled quantum oscillations, │+>⇄ │─>, between near symmetric energy wells, occupying intramolecular decoherence-free subspaces [67, 68, 69]).
Reduced energy product enol and imine protons occupying heteroduplex heterozygote [15, 16, 17, 23] sites G´- C´, *G-*C, *A-*T contain EPR-generated [29, 30, 31], entangled proton qubits [35, 36, 37, 38, 39], shared between two indistinguishable sets of electron lone-pairs [65] belonging to decoherence-free subspaces [11, 66, 67, 68, 69] of enol oxygen and imine nitrogen on opposite genome strands (Figures 1-4).
![Figure 2: (a) Symmetric and (b) asymmetric channels for EPR-generated [29-31] proton-proton separation and electron arrangements at a G-C site.) ((a) Symmetric channel for proton exchange tunneling electron rearrangement, yielding two enol-imine hydrogen bonds between complementary G-C. Here an energetic guanine amino proton initiates the reaction. (b) The asymmetric exchange tunneling channel, yielding the G-C “hybrid state” containing one enol-imine and one keto-amino hydrogen bond. An energetic cytosine amino proton initiates reaction in this channel. An annulus of reaction is identified by arrows within each G-C reactant duplex. Electron lone-pairs are represented by double dots: Subscript notation for *G0200, etc. is given in Figure 3 legend).](/fulltextimages/1462/fig_2.jpeg)
Figure 2: (a) Symmetric and (b) asymmetric channels for EPR-generated [29, 30, 31] proton-proton separation and electron arrangements at a G-C site.) ((a) Symmetric channel for proton exchange tunneling electron rearrangement, yielding two enol-imine hydrogen bonds between complementary G-C. Here an energetic guanine amino proton initiates the reaction. (b) The asymmetric exchange tunneling channel, yielding the G-C “hybrid state” containing one enol-imine and one keto-amino hydrogen bond. An energetic cytosine amino proton initiates reaction in this channel. An annulus of reaction is identified by arrows within each G-C reactant duplex. Electron lone-pairs are represented by double dots: Subscript notation for *G0200, etc. is given in Figure 3 legend).
Consequently, product enol and imine proton qubits
participate in entangled quantum oscillations, │+>⇄ │─>,
at ~ 4×1013 s−1 between near symmetric energy wells
(Figure 20; Table 8-9), in decoherence-free subspaces
[17, 54, 67, 68, 69], until “measured by”, δt << 10–13 s,
Grover’s-type [11, 40] quantum processors [35, 36, 37, 38, 39]. This
creates an entanglement state between measured
“groove” protons [70] and the enzyme quantum processor
[40] that subsequently implements quantuminformation
processing, Δtʹ ≤ 10–14 s [13, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45]. Quantum information
processing measurements of G´-C´and*G-*C sites
(entangled proton qubit states) specify time-dependent
substitutions, ts, exhibited as G′2 0 2 → T, G′0 0 2 → C, *G0
2 00 → A& *C2 0 22 → T (see Table 1 & Figure 5 legend for
notation) whereas, time-dependent deletions, td [16, 17],
are exhibited as *A → deletion and *T → deletion. These
observables [15, 16, 17, 20, 21] are not consistent with
classical [27, 28] transcription and replication, but are
entirely compatible with Grover’s [40] enzyme quantum
processors {see Equation (15)} measuring quantum
informational content [35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45] embodied within EPR-
$$ \text{generated} \quad [29-31] \quad keto-amino \quad -(entanglement) \rightarrow $$
enol−imine ─ entangled enol and imine proton qubits.
(Here distinguish entanglement originated ts, e.g., G′ → T,
from classical Newtonian substitutions, e.g., G → T [27, 28].
Entanglement generated ts, e.g., G′2 0 2 → T [35, 36, 54],
are mechanistically, and therefore biologically,
distinguishable from classical “Muller-type” [71, 72]
substitutions, e.g., Newtonian, G → T [28].
Also, when G′ and/or *C are located on the transcribed
strand of T4 phage heteroduplex heterozygotes, r+/rII
[15, 16, 17, 20, 21], and substitutions, G → T and/or C → T, are
required to express the wild-type r+ allele [23, 73],
standard Watson-Crick DNA growth does not occur on E.
coli K (nonpermissive host) unless the r+ allele (genetic
information) has been transcribed and expressed. In
these cases, the wild-type r+ allele requires expression
(transcription and translation) yielded by classical
biological operations on physical substitutions, i.e., G → T
and/or C → T, before DNA growth can occur on E. coli K
[23, 73]. Curiously, heteroduplex heterozygote T4 phage ts
systems [15, 16, 17, 23] — i.e., time-dependent “point”
mutations, G-C → G´-C´, G-C → *G-*C, A-T → *A-*T, that
accumulate in metabolically inert suspensions of T4
Phage particles [18, 19, 20, 21, 22, 23] — routinely exhibit identical G′ →
T and *C → T mutation frequencies expressed by pre-
replication transcription, and post-transcription replication [16, 17, 20, 21].
Although classical models [27, 28] cannot explain these routine observables [15, 16, 17, 20, 21], identical G′ → T and *C → T mutation frequencies expressed by pre-replication transcription and post-transcription replication are consistent with quantum information processing, Δtʹ ≤ 10–
14 s (see Figure 5), of EPR-generated [29, 30, 31] entangled proton qubits [35, 36, 37, 38, 39], by Grover’s [40] quantum processors. Consequently, classical experimental molecular genetic investigations to provide a rationale for nonclassical, Gʹ → T &*C → T transcription and replication observations [15, 16, 17, 18, 19, 20, 21, 22] were ultimately abandoned [27]. The nonclassical, identical G′ → T and *C → T mutation frequencies, expressed by pre-replication transcription and post-transcription replication [20, 21], are also exhibited by evolving microsatellite [52, 74], short tandem repeats(STRs) within human and rat genomes [35, 38]. These nonclassical observables [20, 21, 27, 38]
![Figure 4: Metastable and entangled proton qubit _*A-*T_ states. (Figure 4: Pathway for metastable keto-amino A-T protons to populate enol and imine proton qubit states. Dashed arrows indicate proton oscillatory pathway for enol and imine proton qubit _*A-*T_ states. Notation is given in Figure 3 legend. The # symbol indicates the position is occupied by ordinary hydrogen unsuitable for hydrogen bonding.) Suggest quantum information processing of EPR- generated entangled proton qubits [35-39] is operational in all duplex DNA and RNA molecular genetic systems [15-22]. This led to identifying EPR-generated entangled proton qubits [35-39,49-51,54] measured by, δt << 10–13 s, Grover’s [40] quantum processors, which form enzyme – proton entanglement states that implement, and execute, quantum information processing, Δtʹ ≤ 10–14 s [13,35-39], before genome duplication is initiated [23]. Consistent with evolution theory [4,28], _ts_ and _td_ properties exhibited by ancient heteroduplex heterozygote T4 phage _ts_ systems [15-17] ― i.e.,_G´-C´_, _*G-_ _*C_& _*A-*T_ base pairs containing EPR-generated [29-31] entangled proton qubits― are also exhibited by (_a_) human rodent STR genomic evolution [38,52], and by (_b_) human gene systems [35-37,39,47-51]. These observations and analyses imply EPR-generated, entangled proton qubits [35-39] occupy decoherence-free subspaces [67-69] for months, years and/or decades, if their measurements, in fact, simulate evolutionary distributions of the 22 most abundant STRs belonging to rat and human genomes [52], which the EPR-entanglement algorithm model does very well [35,38]. In this case, a substantial percentage of EPR- generated entangled proton qubits occupying decoherence-free subspaces [11,37-39,67-69] is “isolated” from their decohering environments [4-6] until “measured by” Grover’s [40] quantum processors [35,36].](/fulltextimages/1462/fig_4.jpeg)
Figure 4: Metastable and entangled proton qubit *A-*T states. (Figure 4: Pathway for metastable keto-amino A-T protons to populate enol and imine proton qubit states. Dashed arrows indicate proton oscillatory pathway for enol and imine proton qubit *A-*T states. Notation is given in Figure 3 legend. The # symbol indicates the position is occupied by ordinary hydrogen unsuitable for hydrogen bonding.) Suggest quantum information processing of EPR- generated entangled proton qubits [35, 36, 37, 38, 39] is operational in all duplex DNA and RNA molecular genetic systems [15, 16, 17, 18, 19, 20, 21, 22]. This led to identifying EPR-generated entangled proton qubits [35, 36, 37, 38, 39, 49, 50, 51, 54] measured by, δt << 10–13 s, Grover’s [40] quantum processors, which form enzyme – proton entanglement states that implement, and execute, quantum information processing, Δtʹ ≤ 10–14 s [13, 35, 36, 37, 38, 39], before genome duplication is initiated [23]. Consistent with evolution theory [4, 28], ts and td properties exhibited by ancient heteroduplex heterozygote T4 phage ts systems [15, 16, 17] ― i.e.,G´-C´, *G- *C& *A-*T base pairs containing EPR-generated [29, 30, 31] entangled proton qubits― are also exhibited by (a) human rodent STR genomic evolution [38, 52], and by (b) human gene systems [35, 36, 37, 39, 47, 48, 49, 50, 51]. These observations and analyses imply EPR-generated, entangled proton qubits [35, 36, 37, 38, 39] occupy decoherence-free subspaces [67, 68, 69] for months, years and/or decades, if their measurements, in fact, simulate evolutionary distributions of the 22 most abundant STRs belonging to rat and human genomes [52], which the EPR-entanglement algorithm model does very well [35, 38]. In this case, a substantial percentage of EPR- generated entangled proton qubits occupying decoherence-free subspaces [11, 37, 38, 39, 67, 68, 69] is “isolated” from their decohering environments [4, 5, 6] until “measured by” Grover’s [40] quantum processors [35, 36].
![Figure 5: Approximate structure “seen by” enzyme quantum processor systems. Figure 5 Approximate proton−electron hydrogen bonding structure “seen by” Grover’s [40] enzyme quantum reader in intervals, δt << 10–13 s, encountering (a) normal thymine, T22 0 22; (b) enzyme-entangled enol- imine _G'2 0 2_; (c) enzyme-entangled imino cytosine, _*C2 0_ _2__2_, and (d) enzyme-entangled enol-imine _G'0 0 2_. Notation is specified in Figure 3 legend. Successful implementation of Grover’s [40] processors executing quantum information processing of EPR- generated [29-31], entangled proton qubits in ancient [46] T4 phage [15-17] and modern eukaryotic [37-39,54] systems argues that “ancient” quantum transcription of entangled proton qubits [35,36], and attendant translation, antedate “standard” classical transcription [28] of genetic information where “evolved” translation [57] is executed in terms of fully developed ribosomes with tRNAs, etc. In this case, ancestral ribozyme – RNA duplex systems [35,36,58-61] acquired rudimentary quantum processing [40] abilities to implement quantum transcription, and attendant translation, of EPR-generated [29-31] entangled proton qubits, and consequently, such ancestral genomic systems would not necessarily be evolutionarily terminal as concluded by Koonin’s [57] classical assessments. Origin of quantum enhanced genetic information required selection of variant primitive RNA – ribozyme peptide systems to execute quantum processing [40-45], which incrementally generated quantum entanglement algorithmic processes that yielded RNA protein systems [35,36], from which DNA protein systems emerged. The ensuing entanglement-enabled genomic evolutionary pathways [36] identify rationale and originsof age-related human disease [35,39], including Huntington’s disease [37,53,54], age-related cancer [35,39,56,75], Alzheimer’s disease [35,39,76,77], and by analogous arguments, ALS (amyotrophic lateral sclerosis [78,79]).“Classical only” molecular genetic assessments of these human maladies have neglected entanglement-enabled contributions [35- 39,54] responsible for evolutionary and molecular genetics manifestations, and consequently, have perpetrated misleading conclusions.](/fulltextimages/1462/fig_5.png)
Figure 5: Approximate structure “seen by” enzyme quantum processor systems. Figure 5 Approximate proton−electron hydrogen bonding structure “seen by” Grover’s [40] enzyme quantum reader in intervals, δt << 10–13 s, encountering (a) normal thymine, T22 0 22; (b) enzyme-entangled enol- imine G'2 0 2; (c) enzyme-entangled imino cytosine, *C2 0 22, and (d) enzyme-entangled enol-imine G'0 0 2. Notation is specified in Figure 3 legend. Successful implementation of Grover’s [40] processors executing quantum information processing of EPR- generated [29, 30, 31], entangled proton qubits in ancient [46] T4 phage [15, 16, 17] and modern eukaryotic [37, 38, 39, 54] systems argues that “ancient” quantum transcription of entangled proton qubits [35, 36], and attendant translation, antedate “standard” classical transcription [28] of genetic information where “evolved” translation [57] is executed in terms of fully developed ribosomes with tRNAs, etc. In this case, ancestral ribozyme – RNA duplex systems [35, 36, 58, 59, 60, 61] acquired rudimentary quantum processing [40] abilities to implement quantum transcription, and attendant translation, of EPR-generated [29, 30, 31] entangled proton qubits, and consequently, such ancestral genomic systems would not necessarily be evolutionarily terminal as concluded by Koonin’s [57] classical assessments. Origin of quantum enhanced genetic information required selection of variant primitive RNA – ribozyme peptide systems to execute quantum processing [40, 41, 42, 43, 44, 45], which incrementally generated quantum entanglement algorithmic processes that yielded RNA protein systems [35, 36], from which DNA protein systems emerged. The ensuing entanglement-enabled genomic evolutionary pathways [36] identify rationale and originsof age-related human disease [35, 39], including Huntington’s disease [37, 53, 54], age-related cancer [35, 39, 56, 75], Alzheimer’s disease [35, 39, 76, 77], and by analogous arguments, ALS (amyotrophic lateral sclerosis [78, 79]).“Classical only” molecular genetic assessments of these human maladies have neglected entanglement-enabled contributions [35, 36, 37, 38, 39, 54] responsible for evolutionary and molecular genetics manifestations, and consequently, have perpetrated misleading conclusions.
| Quantum flip flop States | Allowable Pair Formation at Replication | Transcription Message | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal Bases | Syn-Purines | ||||||||||||||||
| G 22000 | C 00222 | A 002# | T 22022 | G 222# A 002# | |||||||||||||
| G' 002 | G-C → C-G | U | |||||||||||||||
| G' 202 | G-C → T-A | T 220202 | |||||||||||||||
| G' 200 | no detectable | G 22000 | |||||||||||||||
| G' 000 | U | ||||||||||||||||
| *G 020° | G-C → A-T | U | |||||||||||||||
| *G 220° | U | ||||||||||||||||
| C' 220 | U | ||||||||||||||||
| C' 020 | U |
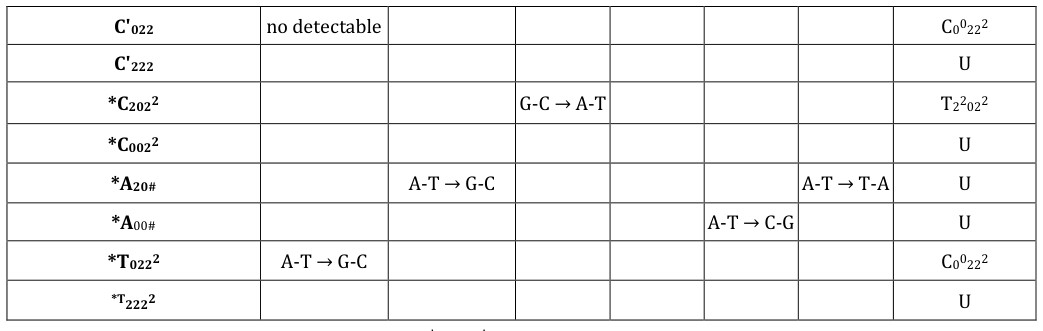
- C'022 no detectable
- C00222
- C'222
- U
- *C2022
- G-C → A-T
- T22022
- *C0022
- U
- *A20#
- A-T → G-C
- A-T → T-A
- U
- *A00#
- A-T → C-G
- U
- *T0222
- A-T → G-C
- C00222
- *T2222
- U
Table 2: Relation between entangled“flip-flop”, │+>⇄│─>,proton qubit states (left column) and transcribed message
This and other reports [37, 38, 39] argue that EPR-generated entangled proton qubits originated inancestral ribozyme – RNA duplex segments. Hence, Grover’s processors were selected “to process” EPR-generated quantum informational content, thereby avoiding evolutionary extinction. In this scenario, measurements on EPR- generated entangled proton qubits have been operational in all duplex genomic systems over the past ~3.6 or so billion y [57, 58, 59, 60, 61]. Misidentification of EPR-generated [29, 30, 31] entangled proton qubits [18, 19, 20, 21, 22, 23, 24, 25], and their subsequent processing by Grover’s-type [40] quantum readers [15, 16, 17, 49, 50, 51], has delayed recognition of quantum information processing of EPR-generated entangled proton qubits [35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49]; classical information processing approximations yield inaccurate results. For example, the quantum entanglement algorithm accurately predicts the evolutionary distributions of the 22 most abundant STRs [38, 52, 74] common to rat and human genomes, which is not available with classical models. These results require significant stability of EPR- generated [29, 30, 31] entangled enol and imine proton qubits, │+>⇄│─>, occupying decoherence-free subspaces [35, 67, 68, 69], until measured by Grover-type [11, 36, 37, 38, 39, 40] quantum processors. Models for origin of life on Earth [57, 58, 59, 60, 61, 62, 63] must provide plausible explanations for (A) origin of self-replicating, sustainable molecular systems and (B) origin of carbon and other volatile elements – nitrogen, sulfur, hydrogen at sufficient concentrations [80, 81] to support earliest ancestral life on Earth’s surface, > ~ 4 billion years ago [60, 82, 83]. The purpose of this report is to provide plausible models for (A) and (B), and therefore, to identify chemical and physical pathways [35, 36, 37, 38, 39] operating within accessible and appropriate prebiotic environments [57, 58, 59, 60, 61, 62, 63] that were exploited to yield origins of earliest biological RNA World [58, 59, 60, 61] life forms, which, consequently, can employ quantum entanglement [7, 8, 9, 10, 11, 37] resources to incrementally “evolve” into sustainable duplex DNA systems (Figure 17) [57, 84]. This pre-LUCA (last universal cellular ancestor [57]) origin of EPR-generated entangled proton qubits, measured by Grover’s-type [40] quantum processors, has enabled genomic evolution to exhibit advances in biological complexity [35], of the form: ribozyme – RNA → genetic code origin → RNA – protein → DNA – protein (Figure 17), over the past ~ 3.6 billion years [36]. Consequences of these entanglement-enabled evolutionary processes [35, 36] identify quantum mechanical rationale and origins of age-related human disease [37, 38, 39].
Origin of Surface Carbon for Earliest RNA – Ribozyme Systems
This origin of life discussion is within the context of a “Big Bang” [85, 86] or “Big Bounce” [87, 88] origin (~13.8 bya, i.e., billion years ago) of mass, particles, energy, and information embedded within massive particles and energy fields (nuclear, gravitational, thermal, and electromagnetic) that specify how particles and energy fields self-interact and interact with each other (Figure 7). About 4.6 bya, Earth and its solar system emerged from a dense solar nebula cloud, schematically depicted in Figure 7 (Nature timeline).
![Figure 7: Nature Timelinespectrum of cosmological processes since the Big Bang [85,86] or Big Bounce [87,88], including origin of life on Earth.](/fulltextimages/1462/fig_7.jpeg)
Current evidence [57, 58] supports the concept that an ancient RNA World [59, 60, 61], i.e., RNA – ribozymes, existed before DNA and protein systems. Fossils indicating living “stromatolites” existed ~ 3.7 billion y ago [83] imply duplex ribozyme – RNA segments existed> ~ 3.9 or so billion years ago [35, 36]. Consistent with Goldman and Taublyn [89], ~ 4.3 to 3.9 billion y ago, asteroids and icy comets containing primordial hydrocarbons [90], long chain polycyclic aromatic hydrocarbons [91, 92, 93, 94, 95], and Fullerenes [96] collided with a cooling prebiotic Earth to create impact reactive environments conducive to formation of complex organic molecules. Since origins of self-replicating “genome-like” polymers require existence of informational molecules necessary to initiate self- replication, one can postulate that synthetic processes described by Goldman and Taublyn [89] could have participated in generating precursors for amino acids, polypeptides, RNA, DNA and small-chain aromatic hydrocarbons, including short “RNA-like” polymers [58, 59, 60, 61]. Combinations of reactive products could incrementally become selectively advantageous for the creation of molecular complexes to synergistically add or incorporate analogous molecular units, and implement primitive polymerizations of nucleotides, oligomers and peptides [60, 61]. In these cases, advantageous reactive processes were preferentially selected by environmental conditions. Over a period of ~ 300×106 y, impact synthetic processes generated “ribozyme-like” RNA polymers, from which primitive, but functional, “ribozyme-like” [61] structures emerged. Primordial molecular polymer complexes on prebiotic Earth [57, 58, 59, 60] could generate probabilistic variant systems that occasionally would exhibit improved efficiencies at surviving in their environments. These incremental classical [12, 28] improvements allowed “original” molecular complexes to acquire “RNA-like” polymer structures, e.g., ribozymes [57, 60] which can inefficiently duplicate ~ 10 to 80 or more molecular RNA units per 24 hrs. This nebulous explanation provides a scenario for possible origin of ancient ribozymes [61] to have emerged ~ 4.1 billion years ago (Figure 7) in primordial pools, continually impacted by icy comets [89]. The present discussion argues that earliest molecular “life forms” [60, 82] precursor RNA – ribozyme polymer segments emerged ~ 4 bya (Figure 7) in primordial pools, continually impacted by icy comets [89]. This scenario requires an explanation for existence of sufficient carbon atoms on Earth’s surface to support carbon-based life, since volatile elements like carbon, nitrogen, sulfur and hydrogen would have vaporized into space, or bonded with iron-rich alloys before precipitating into Earth’s metallic core [80, 81]. Recent studies by Dasgupta and colleagues [97] imply that Earth’s carbon could have originated from a collision with a Mercury-like planet embryo around 4.4 billion years ago. This collision, illustrated in Figure 8-9, could have captured and preserved sufficient carbon atoms on Earth’s surface, from which carbon-based life could have emerged.
![Figure 8: The ratio of volatile elements in Earth’s mantle suggests that virtually all of the planet’s life-giving carbon came from a collision with an embryonic planet approximately 100 million years after Earth formed [97]. (Credit: Image by A. Passwaters/Rice University based on original courtesy of _NASA/JPL-Caltech_](/fulltextimages/1462/fig_8.jpeg)
![Figure 9: Schematic of proto-Earth’s merger with a potentially Mercury-like planetary embryo, a scenario supported by new high pressure-temperature experiments at Rice University. Magma ocean processes could lead planetary embryos to develop silicon- or sulfur-rich metallic cores and carbon-rich outer layers. If Earth merged with such a planet early in its history, this could explain how Earth acquired its carbon and sulfur. (Credit: Figure courtesy of Rajdeep Dasgupta [97] - See more at: http://news.rice.edu/2016/09/05/study-earths-carbon- points-to-planetary smashup/#sthash.gnVASfTj.dpuf](/fulltextimages/1462/fig_9.jpeg)
Figure 9: Schematic of proto-Earth’s merger with a potentially Mercury-like planetary embryo, a scenario supported by new high pressure-temperature experiments at Rice University. Magma ocean processes could lead planetary embryos to develop silicon- or sulfur-rich metallic cores and carbon-rich outer layers. If Earth merged with such a planet early in its history, this could explain how Earth acquired its carbon and sulfur. (Credit: Figure courtesy of Rajdeep Dasgupta [97] - See more at: http://news.rice.edu/2016/09/05/study-earths-carbon- points-to-planetary smashup/#sthash.gnVASfTj.dpuf
Evidence Requiring Entanglement-State Quantum Transcription
The bacteriophage T4 genome contains about 130 genes within 168,903 bp [46], and it also contains an rII region of ~1600 bp [23, 73] that is exploited for fine scale genetic mapping of time-dependent “point” base substitutions, ts [17], and deletions, td [16], with base pair resolution. When T4 phage infects a nonpermissive host (e.g., Escherichia coli K), the wild-type r+ allele must be transcribed and expressed, i.e., translated after transcription, before DNA replication is initiated [23, 73]; otherwise, T4 phage is not capable of growth on a nonpermissive host, E.coli K. In studies of time-dependent rII →r+mutations exhibited by bacteriophage T4 [15, 16, 17, 20, 21, 54], a mutant base pair is substituted at one of the 300 or so mapped genetic sites in rII region DNA, thereby eliminating wild-type r+ alleles. When T4 phage rII mutant systems require substitutions, G → T or C → T, to express the r+ allele, growth does not occur on E. coli K unless the relevant “point” base substitution, G → T or C → T, has been implemented [23, 73]. However, when the relevant rIIG-C mutation site exhibits heteroduplex heterozygote conditions ―G′-C′ or_*G-*C_sites populated by EPR-generated [29, 30, 31] entangled proton qubits ― growth on E. coli K is allowed [23, 54]. In these cases,Grover’s-type [40] quantum transcription of entangled proton qubits at G′-C′ or *G-*C sites (Figure 5) is “deciphered”, i.e., translated, to determine the answer to an observable feedback loop question – “Yes” or “No” – regarding initiation of genome duplication. In cases of T4 phage infecting E. coli K [23, 73], if operational information generated by quantum transcription [35, 36, 37, 38, 39] – e.g., G′ → T or *C → T (Figure 5) – communicates existence of the r+ allele, replication is subsequently initiated before physical substitutions, G′ → T or *C → T, are incorporated [15, 16, 17]. In these situations, confirmation of the r+ allele is provided before replication initiation by enzyme quantum reader measurements of entangled proton qubit G′-C′ or *G-*C states (Figure 5). The molecular base sequence identifying the r+ allele does not physically exist until accurate replication subsequently introduces physical substitutions, G′ → T or *C → T, which does not occur unless r+ information was previously communicated by quantum (a) transcription, (b) translation and (c) positive feedback loop specificity, confirming existence of an operational r+ allele [16, 17, 35, 36]. In these cases, a base sequence specifying the r+ allele did not physically exist, but r+ information was confirmed by translation of quantum information generated by Grover’s-type quantum transcription of EPR-generated entangled proton qubits occupying G′-C′ or *G-*C superpositions (Figure 5). Therefore, observable “measurements” [16, 17, 20, 21, 54] on entangled proton qubit states occupying ancient T4 phage DNA imply quantum transcription, and attendant translation, antedate “standard” transcription of keto-amino states where “evolved” translation is executed in terms of fully developed ribosomes with tRNAs, etc. [28, 57]. In this case, availability of entangled proton qubit states in ancestral RNA – ribozyme systems [35, 36] could allow primordial quantum transcription and attendant translation processes to incrementally introduce increases in fitness by exploiting quantum informational content and reactive properties of entangled proton qubits [11, 37, 38, 39], thereby incrementally generating DNA – protein systems from ancestral RNA – ribozyme systems. Thus RNA ribozyme systems [36] would not necessarily be evolutionarily terminal as concluded by Koonin’s [57] classical assessments.
Quantum Entanglement Algorithm Applications
General Properties
The quantum entanglement algorithm [35] for implementing quantum information processing [40, 41, 42, 43, 44, 45] of EPR-generated [29, 30, 31] entangled proton qubits [36] is specified as follows. Hydrogen bonding amino protons within standard G-C and A-T base pair segments are subjected to quantum uncertainty limits [2, 66], Δx Δpx ≥ ћ/2, which cause direct quantum mechanical proton – proton physical interaction in confined spaces, Δx [98, 99]. This generates probabilities of EPR arrangements [29, 30, 31], keto-amino ― (entanglement) → enol-imine, observed as [16, 17] G-C → G´-C´, G-C → *G-*C and A-T → *A-*T (see Figure 1-5 for notation; e.g., G´-C´, are used to denote necessity of Hilbert space to describe entangled proton qubit dynamics [35, 43]). In these EPR reactions, position and momentum entanglement [7, 8, 9, 10] is introduced between separating enol and imine protons [35, 36, 37, 38, 39]. Reduced energy product enol and imine protons are shared between two intramolecular sets of indistinguishable electron lone-pairs, belonging to enol oxygen and imine nitrogen, in decoherence-free subspaces [11, 67, 68, 69] on opposite genome strands, and thus, participate in entangled quantum oscillations [17, 39] between near symmetric energy wells at ~ 4×1013 s−1(Table 8-9, Appendix II) until “measured by”, δt << 10–
13 s, Grover’s-type [40] quantum processor. This creates an entanglement state between measured “groove” proton(s) [70] and Grover’s enzyme processor.
This proton – processor entanglement yields time- dependent,molecular clock [100, 101, 102] substitutions, ts, and time-dependent deletions [16], td, after quantum information processing, Δtʹ ≤ 10–14 s [13, 35, 36, 37, 38, 39], events of (i) quantum transcription, (ii) translation, (iii) selection of accessible amino acids for peptide bond formation, (iv) random genetic drift [103], and (v) initiation of genome growth (see Table 2). Energy for peptide bond formation ~ 8 to 16 KJ/mole [28] is provided by decoherence of proton – processor entanglement [35, 36, 37, 38, 39]. Time- dependent substitutions, ts, are exhibited as G′2 0 2 → T, G′0 0 2 → C, *G0 2 00 → A_and *C2 0 2_2 → T, which are expressed as EPR-generated SNPs [15, 16, 17, 23, 35, 36, 37, 38, 39, 100, 101, 102], whereas td are consequences of *A-*T site deletions (Figure 4). The enzyme quantum reader distinguishes between EPR-generated ts, identified above, and classically originated Muller-type [28, 71] SNPs (see Table 2). For example, cancer-causing “driver mutations” [50, 55, 75] are associated with EPR-originated ts [35, 39], whereas “passenger mutations” [55] are classically originated SNPs [35, 39]. Additionally, ts and td can introduce and eliminate initiation codons UUG, CUG, AUG, GUG and termination codons UAG, UGA, UAA which introduces variable clock “tic-rates” [15, 16, 17, 35, 100, 101, 102, 103], and allows entanglement- enabled genomic growth via “expansions” [104, 105]. In duplex DNA of human genomes, unstable repeats [53, 106, 107, 108, 109, 110, 111] exhibit expansions and contractions via dynamic mutations [37, 38, 39, 104, 105], where (CAG)n sequences (n > 36) [109] can exhibit expansions ≥ 10 (CAG) repeats in 20 y [37, 106, 109]. This observation implies the hypothesis that susceptible ancestral genomes implemented EPR- dynamic mutation expansions as consequences of specific ts [15, 16, 17, 36]. A “net” triplet repeat dynamic mutation expansion rate of 13 repeats, e.g., (CAG)13 = 39 bp, per 20 y for 3.5 billion y would generate a genome of ~ 6.8×109 bp, which is “ballpark” compatible with bp content of the Homo sapiens genome [28, 57]. Based on the present and previous studies [37, 38, 39, 54, 104], evolutionary genomic growth was, and is, a consequence of the EPR-generated [29] quantum entanglement algorithm [35, 36] introducing, and eliminating, initiation codons ─ UUG, CUG, AUG, GUG ─ and stop codons, UAA, UAG & UGA [37, 104]. This hypothesis is consistent with the fact that overall microsatellite content in a genome correlates with genome size of the prokaryotic or eukaryotic organism [112]. Selected “expansion” sequences were exploited as conserved genes, e.g. [37, 75, 76, 77, 78, 79], whereas “other” expansion sequences have been relegated to “unspecified” conserved noncoding genomic space (CNGS) [113, 114].
The quantum entanglement algorithm [36] model for genome growth is tested by correctly predicting the evolutionary distribution of the 22 most abundant microsatellites (short tandem repeats, STRs [52]) common to human and rat genomes [35, 38]. Although this STR evolutionary distribution is, classically, an unresolved enigma [52, 74, 112], its analyses in terms of quantum entanglement algorithm predictions [37, 38] agree with observation [52]. This agreement implies origins of (a) double helix DNA components [36, 37], (b) conserved noncoding genomic spaces (CNGS) [113, 114] and (c) corresponding STRs [52, 74], all of whichcan be explained in terms of quantum entanglement evolutionary dynamics [35, 36, 37, 38, 39]. Additionally, accuracy of EPR-generated entangled proton qubit analyses of the evolutionary distributions of the 22 most abundant STRs [38, 52] common to rat and human genomes requires a significant percentage of EPR-generated entangled enol and imine proton qubits, │+>⇄│─>, to remain stable for months, years to decades, until quantum information ― measured by Grover’s [40] quantum processors ―is evolutionarily perpetrated. Otherwise, these quantum entanglement analyses [35] would be inaccurate, which is contrary to fact [37, 38, 52].
Origin and Implementation of Quantum Entanglement Information Processing
Quantum information processing exhibited by ancient [46] T4 phage DNA [15, 16, 17, 35], and human gene systems [37, 38, 39, 49, 50, 54] falsify the in vivo_anti-entanglement hypothesis [5, 6], and require an evolutionary origin [36]. This and previous reports [16, 17, 37, 38, 39] argue that “earliest” quantum information processing was selected by duplex segments of ancestral proto-RNA – ribozyme molecular complexes [35, 36, 57, 58, 59, 60, 61]. In this situation, random classical processes [12] introduced energetically preferable hydrogen bonded base pairs [65] between metabolically inert [64], complementary RNA – ribozyme duplex segments. Consequently, quantum uncertainty limits [2, 66], Δx Δpx ≥ ħ/2,operate on metastable hydrogen bonding amino (−NH2) protons [65] to introduce probabilities of EPR [28, 29, 30, 31] arrangements, _keto-amino ―(entanglement)→ enol−imine. Each reduced energy, entangled imine and enol product proton is shared between two indistinguishable sets of electron lone-pairs (Figure 1-4), and therefore, participates in entangled quantum oscillations [35, 36, 37, 38, 39] at ~ 4×1013 s−1 (Figure 3, Table 2), into and out of major (~22 Å) and minor (~12 Å) genome grooves [70], between near symmetric energy wells in decoherence- free subspaces [11, 67, 68, 69]. This specifies quantum dynamics of an EPR pair of entangled enol and imine proton qubits until measured, $\delta t << 10^{-13}$ s, by an enzyme quantum processor [38, 39, 40]. The imine and enol protons constitute a pair of entangled two-state proton qubits on opposite genome strands. An entangled enol or imine proton is in state $|+ \rangle$ when it is in position to participate in interstrandhydrogen bonding, and is in state $|- \rangle$ when it is "outside", in a major or minor DNA groove [15, 16, 17, 36, 70]. The quantum mechanical state of the entangled pair of $*G'*C$ proton qubits can be viewed as a vector in the four-dimensional Hilbert space that describes the quantum position state of two protons. The most general quantum mechanical state of these two protons can be written as [98]
$$|\Psi> = C_{++} |++ > +C_{+-} |+-> +C_{--} |+-> C_{--} |+-> (1)$$
where the first symbol, + or -, represents proton 1 and the second symbol represents proton 2, and the expansion coefficients, $c_s$, satisfy normalization, $|c_{++}|^2 + |c_{+-}|^2 + |c_{--}|^2 = 1$. Since Eq (1) cannot be expressed as a tensor product of protons 1 and 2, maximally entangled quantum states for the qubit pair of imine and enol protons can be written in terms of the four Bell [32, 33] states, expressed as
$$|\Phi^+| = 1/\sqrt{2} \{ |++ > +|-> \} (2)$$
$$|\Phi^-| = 1/\sqrt{2} \{ |++ > -|-> \} (3)$$
$$|\varphi^+| = 1/\sqrt{2} \{ |++ > +|-> \} (4)$$
$$|\varphi^-| = 1/\sqrt{2} \{ |++ > -|-> \} (5)$$
The dimensionality of the Hilbert space required to express the quantum mechanical state for four proton qubits occupying $G'-C'$ isomer pair superpositions is sixteen, i.e., $2^N = 2^4 = 16$. Each entangled imine and enol proton is shared between two sets of indistinguishable electron lone-pairs (Figure 1), and thus, participates in entangled quantum oscillations between near symmetric energy wells at $\sim 10^{13}$ s$^{-1}$ in decoherence-free subspaces [35, 36, 67, 68, 69], which specifies entangled proton qubit dynamics occupying a heteroduplex heterozygote $G'-C'$ superposition site [16, 17, 37, 38, 39]. In this case, two sets of entangled imine and enol proton qubits four protons constituting two sets of entangled "qubit pairs" occupy complementary $G'-C'$ superposition isomers such that enzyme quantum reader "measurement" of $G'$-protons specifies, instantaneously [29, 30, 31], quantum states of the four entangled qubits that occupy the sixteen-dimensional space.
Studies of heteroduplex heterozygote $G'-C'$ sites [23], with $G'$ on the transcribed strand [16, 17, 35, 54], require the enzyme quantum reader to "measure", specify and execute quantum informational content of sixteen different entangled proton qubit $G'-C'$ states (Table 2). In the case of Figure 5, $G'0 0 2$ ($G'0 0 2 \rightarrow C$, Table I), the carbon-2 imine proton is in state $|- \rangle$ groove position, whereas the eigenstate $G'2 0 2$ ($G'2 0 2 \rightarrow T$, Table I) has both carbon-2 imine and carbon-6 enol protons in state $|- \rangle$ groove positions (see Figure 5). Eigenstate $G'2 0 0$ ($G'2 0 0 \rightarrow G$; "null" mutation) has the carbon-6 enol proton "trapped" in a state $|- \rangle$ DNA groove, but entangled enol and imine protons for eigenstate $G'0 0 0$ are both in state $|+ \rangle$, the "interior" interstrand hydrogen bond position. Since the enol and imine quantum protons on $G'$ are one-half of the four entangled imine and enol $G'$-proton qubit pairs, enzyme quantum reader measurements on $G'$-proton states specifically select quantum mechanical qubit states, $|- \rangle$ and $|+ \rangle$, for the four entangled $G'-C'$ protons.
Here the entangled pair $-$ guanine carbon-2 imine and cytosine carbon-2 enol are identified, respectively, as proton numbers I and II (Roman numerals). Proton numbers III and IV, respectively, are cytosine carbon-6 imine and guanine carbon-6 enol. Using this notation, the enzyme quantum reader measures the four entangled proton qubit states of $G'0 0 2$ as $|- \rangle$, guanine imine proton I is in state $|- \rangle$, cytosine enol proton II is in state $|+ \rangle$, cytosine imine proton III is in state $|- \rangle$, and guanine enol proton IV is in state $|+ \rangle$. Similarly, the measured proton qubit state for $G'2 0 2$ is $|- \rangle$, and is $|+ \rangle$ for $G'2 0 0$, and finally, is $|+ \rangle$ for eigenstate $G'0 0 0$. In addition to the four quantum mechanical states of $G'$ imposed by enzyme quantum reader measurements (Figure 5b-e), twelve additional states are required to specify the four two-state quantum mechanical proton qubits. The $G'-C'$ site superposition consists of two sets of intramolecular entangled proton qubit-pairs that are participating in quantum oscillations between near symmetric energy wells in decoherence-free subspaces [16, 17, 35, 36, 37, 38, 39, 67, 68, 69] at $\sim 10^{13}$ s$^{-1}$ s. Therefore, the most general quantum mechanical state of these four $G'-C'$ protons is given by [7, 10]
where the $c_i$'s represent, generally complex, expansion coefficients. Since the 16-state superposition of four entangled proton qubits occupy enol and imine "intraatomic" subspaces, shared between two indistinguishable sets of electron lone pairs, the entangled quantum superposition system will persist in evolutionarily selected decoherence-free subspaces [11, 15, 16, 17, 35, 36, 37, 38, 39, 67, 68, 69] until an invasive perturbation, e.g., "measurement" [40], exposes the previously "undisturbed" quantum mechanical superposition [99].
Just before enzyme quantum reader measurement of a $G'$-$C'$ site where $G'$ is on the transcribed strand, the 16-state $G'$-$C'$ superposition system is described by Equation (6). In an interval $\delta t < 10^{-13}$ s, the enzyme quantum reader simultaneously detects entangled $G'$-protons I (carbon-2 imine) and IV (carbon-6 enol) in either correlated position states, $|->$ or $|+>$, which are components of an entangled proton "qubit pair". When proton I or IV is measured by the quantum reader in position state, $|->$ or $|+>$, the other member of this entangled pair will, instantaneously [29, 30, 31], be in the appropriately correlated state, $|+>$ or $|->$, respectively. Protons detected in state $|->$, "outside" groove position [70], form "new" entanglement states with the proximal quantum reader [15, 16, 17, 35, 36, 37, 38, 39, 54] that enable enzyme quantum coherence to implement its quantum search, $\Delta t \leq 10^{-14}$ s, which specifies an incoming electron lone-pair, or amino proton, belonging to the tautomer selected for creating the "correct" complementary mispair (Figure 6). Protons detected in state $|+>$, "inside" hydrogen bonding position, contribute to specificity of the $G'$ genetic code, exemplified by both $G'2$ 0 2 and $*G2$ 0 22 "measured as" normal $T2^2$ 0 22 (Figure 5) via quantum transcription and replication [36, 40]. Since the quantum reader detects entangled $G'$-protons I and IV in states $|->$ or $|+>$, the "matching" correlated quantum states, $|+>$ or $|->$, of entangled $C'$-protons II and III were instantaneously specified.
Consequently, enzyme quantum reader "measurement" on $G'$-protons I and IV converts, instantaneously, the 16-state quantum system of Equation (6) into the 4-state system $c_1|-++>$, $c_5|-++>$, $c_9|-++>$, $c_{13}|-++>$ listed in column B of Table 2 and illustrated in Figure 5b-e, where expansion coefficients, $c_6$ are defined by $c_1 = \sum_{i=1}^{4} c_i$, $c_5 = \sum_{i=5}^{8} c_i$, $c_9 = \sum_{i=9}^{12} c_i$, and $c_{13} = \sum_{i=13}^{16} c_i$. This result is displayed in Table 2 where column A identifies the unperturbed 16-state quantum system of Equation (6). Column B contains the distribution of $|->$ and $|+>$ proton states for $G'$-$C'$ protons: I, II, III, IV generated instantaneously as a consequence of the quantum reader initially "measuring" quantum states of entangled $G'$-protons I and IV. The instantaneously generated quantum states $c_1|-++>$, $c_5|-++>$, $c_9|-++>$, $c_{13}|-++>$ provide, instantaneously, specific instructions for the enzyme proton entanglement before it embarks on its entangled enzyme "quantum quest", $\Delta t \leq 10^{-14}$ s, of selecting the incoming tautomer specified by molecular evolution, $ts$ requirements [16, 17, 36]. Incoming tautomers selected by entangled enzyme quantum searches are identified in column C and resultant molecular clock substitutions, $ts$, are listed in column D of Table 2.
In intervals, $\delta t < 10^{-13}$ s, the enzyme quantum processor measurement apparatus "traps" entangled $G'$ imine and/or enol protons — I and IV — in DNA grooves, specified by state $|->$ and consequently, the position state, $|->$ or $|+>$, is instantaneously specified for the four entangled $G'$-$C'$ protons: I, IV and II, III. In column A of Table 2, an entanglement state between the quantum reader and a "groove" proton is indicated by superscript, "*", e.g., $*-++>$, identifies $G'$ proton I as the enzyme – entangled $G'$-groove proton. The "new" entanglement state between the quantum reader and the "trapped" proton enables enzyme quantum coherence to be immediately exploited in implementing an entangled enzyme quantum search, $\Delta t \leq 10^{-14}$ s, which ultimately specifies the particular $ts$ as $G'0$ 0 2 → C, $G'2$ 0 2 → T or $G'2$ 0 0 → G (Table I). The specificity of each tsis governed by the entangled enzyme quantum search selects the correct incoming tautomers — syn-$G2^2$ 2 #, syn-$A0^0$ 2 #, $C0^0$ 2 22 — respectively, for proton qubit eigenstates — $G'0$ 0 2, $G'2$ 0 2, $G'2$ 0 0 — illustrated.
| A | B | C | D | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| c │*−+−+> 1 c │*−−−+> 2 c │*−−++> 3 c │*−+++> 4 | c │−+−+> 2 c │−+−+> 2 c │−+−+> 3 c │−+−+> 4 | syn-G22 2 # | G′0 0 2 → C | ||||||||
| c │*−++*−> 5 c │*−−−*−> 6 c │*−+−*−> 7 c │*−−+*−> 8 | c │−++−> 5 c │−++−> 6 c │−++−> 7 c │−++−> 8 | syn-A00 2 # | G′2 0 2 → T | ||||||||
| c │+−+*−> 9 c │+++*−> 10 c │++−*−> 11 c │+−−*−> 12 | c │+−+−> 9 c │+−+−> 10 c │+−+−> 11 c │+−+−> 12 | C00 2 22 | G′2 0 0 → G | ||||||||
| c │+−−+> 13 c │++++> 14 c │+−++> 15 c │++−+> 16 | c │+−−+> 13 c │+−−+> 14 c │+−−+> 15 c │+−−+> 16 | none | G′0 0 0 → ? ?= microcolony |
Table 3: Evolution of the sixteen-state entangled proton qubit G´-C´ superposition, before measurement (column A), after measurem
Table 2: Evolution of the sixteen-state entangled proton qubit G´-C´ superposition, before measurement (column A), after measurement, Δt´ ≤ 10−14 s (column B), and decohered observables (column D). (Unperturbed (A) and instantaneous yield of “measured” (B) G′-C′ entangled proton qubit states, showing results of entangled enzyme quantum search, Δt′ ≤ 10_−14_ s, (C) and molecular clock (D) observable results, ts.) In Figure 5, Table 1 and Table 2, Natural selection has exploited quantum entanglement properties of EPR proton qubits [15, 16, 17, 35, 36], which allow enzyme – proton entanglement to specify, and implement, results of an entangled enzyme quantum search in intervals, Δt′ ≤ 10_−14_s [13, 35, 36, 37, 38, 39]. This mechanism implies that enzyme proton entanglement implementation of an enzyme quantum search would not be successful without instantaneous specification [29, 30, 31] of the four G′-C′ entangled proton qubit states determined by quantum reader “measurements” on the two G′-proton qubits, I and IV, associated with the transcribed strand (Table 2).
Enzyme – Proton Entanglement Quantum Search, Δt ≤ 10─ 14 s, Mechanism
The enzyme quantum reader “measurement apparatus” [35, 36] patrols the double helix along major (~ 22 Å) and minor (~ 12 Å) grooves [70], creating entanglement states between “measured” enol and imine entangled qubit “groove protons” and proximal enzyme components [35, 36, 37, 38, 39]. The quantum reader polymerase energy source is ATP, and it maintains a reservoir of purines, pyrimidines and nucleotides for base pairing operations. Davies [115] has noted that the polymerase protein has a mass of about 10−19 g, and a length of about 10−3 cm and travels at a speed of about 100 bp per sec., or about 10−5 cm s−1 [28, 116]. Curiously, the normal speed of the polymerase, ~ 10−5 cm s−1, corresponds to the limiting speed allowed by the energy-time uncertainty relation for the operation of a quantum clock. For a clock of mass m and size l, Wigner [117] found the relation
2 / T ml $$ \mathrm {E} = \mathrm {E} _ {1} + \mathrm {E} _ {2} + \dots + \mathrm {E} _ {n} $$ (7) Equation (7) can be expressed in terms of a velocity inequality given by $$ \mathrm {E} = \frac {1}{2} \mathrm {A} ^ {2} + \mathrm {B} ^ {2} $$ / v ml (8) which, for this polymerase, yields a minimum velocity of about 10−5 cm s−1, implying the quantum reader enzymespeed of operation can be confined by a form of quantum synchronization uncertainty [115]. The quantum reader “measurement apparatus” has been evolutionarily selected to decipher, process and exploit informational content within DNA base pairs composed of either (a) the classical keto-amino state, (b) undisturbed, enol and imine entangled proton qubit states Eqs (2 – 6) including enzyme – proton entanglements participating in an entangled enzyme quantum search, Δt´ ≤ 10_−14_ s [13, 35, 36, 37, 38, 39]. The enzyme quantum measurement-operator is identified by Μ, and operates on G′-proton states located on the transcribed strand to yield three different entanglement states between groove protons and enzyme components. From column B of Table 2, these enzymatic quantum “measurements”, and resulting enzyme-proton entanglements, can be symbolized by [36]
Μ│−+−+ > = ć1│−+−+ >ÊpI (9) Μ│−++− > = ć5│−++− >ÊpI, pIV (10) Μ│+−+− > = ć9│+−+− >ÊpIV, (11) where ÊpI, pIV in Eq (10) represents quantum entanglement between “groove” proton I (G′2 0 2-imine) and “groove” proton IV (G′2 0 2-enol) and proximal enzyme components. Similarly, ÊpI_and Êp_IV , represent alternative entanglements between enzyme components and entangled proton I, and separately, entangled proton IV, respectively. The original unperturbed groove proton “quantumness” becomes distributed over an enzyme “entanglement site”, which is selected to complete its assignment of specifying the complementary mispair before proton decoherence, i.e., Δt′ < τD< 10−13 s [13, 35, 36, 37, 38, 39]. Each of the three enzyme-proton entanglements implements a different “selective” quantum search, Δt′ ≤ 10_−14_ s [36], to specify the correct evolutionarily required purine or pyrimidine tautomer to properly complete the molecular clock [15, 16, 17, 18, 19, 20, 21, 22, 35, 36, 37, 38, 39, 100, 101, 102] base substitution, ts, by a quantum processing [36, 40],Topal-Fresco [16, 17, 72] substitution-replication mechanism (Table 1, Figure 6). Since quantum informational content is deciphered by enzymatic processing of entangled proton qubits occupying decoherence-free subspaces [67, 68, 69] shared between two indistinguishable sets of electron lone-pairs, the entangled enzyme quantum search mechanism [35, 36, 37, 38, 39] initially selects the incoming tautomer based on electron lone-pair, or amino proton, availability. Evidently the “evolved” quantum reader has an immediately accessible “reservoir” of required tautomers for quantum search selection [36]. Evidence discussed here [15, 16, 17, 18, 19, 20, 21, 22, 23] implies an enzyme- entanglement complex has been evolutionarily selected and refined over the past ~ 3.5 or so billion y [35, 36] to implement an entangled enzyme quantum search that interfaces with decoherence-free subspaces [36, 67, 68, 69]. In this model of genomic evolution, an evolutionarily selected enzyme-proton entanglement implements a quantum search of the evolutionarily available purine and pyrimidine database for the “matching” classical tautomer required to execute an “in progress” complementary mispair formation before proton decoherence [13, 35, 36, 37, 38, 39]. The initial component of the complementary mispair the specific eigenstate was selected by “new” quantum entanglement between the “trapped” entangled groove proton and the enzyme quantum reader (Table 2). The enzyme – proton entanglement implements a quantum search which specifies ─ in intervals, Δt′ ≤ 10_−14_ s [13, 35, 36, 37, 38, 39] the incoming electron lone-pair, or amino proton, belonging to the tautomer required to create the complementary mispair (Figure 6, Table 1). This allowed quantum coherence of the entangled ribozyme and/or enzyme to specify the selected ts or td, and thus, enable entanglement-directed genomic evolution [35, 36, 37, 38, 39, 100]. The entanglement-enabled introduction of base substitutions, ts [39, 100], and deletions, td [16, 17, 38], can introduce and eliminate initiation codons UUG, CUG, AUG, GUG and/or stop codons: UAA, UGA, UAG [28]. The resulting “dynamic mutations” [37, 54, 104, 105] can cause unstable (CAG)n (n > 36) repeats to exhibit deletions and/or expansions ≥ 10 (CAG) repeats in 20 y. This mechanism qualitatively predicts the evolutionary expansion and contraction molecular dynamics exhibited by Huntington’s disease (CAG)n repeats [37, 53, 54], and therefore, provides a model for genomic growth from pre-prokaryotic primordial RNA systems,to eukaryotic DNA of Homo sapiensʹ dimensions, ~ 6.8×109 bp, over the past ~ 3.5 billion y [35, 36, 37, 38, 39].
Triplet Code Origin via Entanglement Resource Hypothesis
Evidence [15, 16, 17, 57, 58, 59, 60] and the model [29, 30, 31, 35, 36, 37, 38, 39]
discussed here imply entangled proton qubit resources
were initially introduced into ancestral duplex “RNA-like”
segments associated with primitive RNA–ribozymesystem
[60, 61]. This model further postulates that duplex RNA
segments were selected from the primordial pool [57, 58, 59]
by quantum bioprocesses, operating on entangled proton
qubits, creating peptide – ribozyme – proton RNA
entanglements [35, 36]. Since quantum bioprocessors
“measure” quantum informational content by selecting
entangled proton qubit states, in intervals δt << 10−13 s
[13, 35, 36, 37, 38, 39], quantum reader operations can be
approximated by a “truncated” Grover’s [39, 40] quantum
search of “susceptible” proton qubits occupying G′-5HMC′
$$ \text {a n d} ^ {* G - 5 H M ^ {*} C (5 H M C} = $$
5hydroxymethylcytosine)superposition sites [36, 46].
Grover's algorithm [40] is applicable for large system
sizes N in high dimensional Hilbert spaces where the
quantum-enabled database is unsorted. However, a
quantum bioprocessor searching an unsorted database of
N qubit states (here N = 20 qubit states occupying G′-C′ +
*G-*C sites) could be approximated by successive
iterations of a “truncated” Grover’s [40] quantum search.
The quantum bioprocessor is designed to identify
entangled proton qubit states, including those occupying a RNA groove, where the "measurement" interval satisfies, $\delta t < 10^{-13}$ s. The quantum bioprocessor peptide–ribozyme forms an entanglement state with the "trapped" proton (Table 2) that, before proton decoherence, $t_0 < 10^{-13}$ s, $(a)$ generates quantum transcription from "measured" entangled proton qubit states [15, 16, 17, 54], e.g., $G'2 0 2 \rightarrow U, 5HMC'2 0 2^2 \rightarrow U$, etc., $(b)$ implements a "new" peptide bond between an "incoming" selected amino acid and an existing "in place" amino acid, and $(c)$ implements selection of an "incoming" tautomer to "pair with" the decohered eigenstate, specified by the "trapped" proton (Table 2) in a genome groove [35, 36, 37, 38, 39, 70].
Quantum bioprocessor operations can therefore be qualitatively approximated by a "truncated" Grover's algorithm [40]. This representation of a quantum bioprocessor's measurement on entangled proton qubit states occupying $G'-5HMC'$ and $*G-5HMC'$ superpositions implies a "truncated" $(N = 20$ qubit states) Grover's [40] algorithm would yield an improved efficiency of $\sqrt{N}$ over a classical search. If $J$ is the total number of bio-molecular quantum reader measuring operations, Grover's "truncated" algorithm states,
$$(2J+1)\arcsin\left(\frac{1}{\sqrt{N}}\right) = \frac{\pi}{2} \quad (12)$$
which yields the interesting solutions,
$$J = 1, \quad N = 4 \quad (13)$$
$$J = 2, \quad N = 10.4 \quad (14)$$
$$J = 3, \quad N = 20.2 \quad (15)$$
$$J = 4, \quad N = 33.2 \quad (16)$$
Consistent with observables exhibited by T4 phage DNA [16, 17], the model outlined here assumes quantum reader measurements of $G'-5HMC'$ and $*G-5HMC'$ superpositions generated RNA "transcription qubits" (Table 1) $G'2 0 2 \rightarrow U, G'0 0 2 \rightarrow 5HMC, *G0 2 0^2 \rightarrow A, 5HM*C2 0 2^2 \rightarrow U$ that provided single base RNA informational units as precursor mRNA and precursor tRNA. (Here ancestral RNA – ribozyme duplex segments are assumed to have been composed of analogs of G – 5HMC and A – U [28, 46]).
Measurements [16, 17, 54] imply $*C2 0 2^2 \rightarrow T$generates $*G0 2 0^0 \rightarrow A$ (~100%) in the complementary strand. Precursor tRNA components were evidently retained in the bio-molecular quantum processor's "hard drive" reservoir until a sufficient "sampling" of entangled qubit states had been subjected to the selected set of measurements. In this case, the number of measurement operations, $J$, converged to a value that yielded adequate statistics. This qualitative model implies the quantum entanglement algorithm, implemented by ribozyme peptide quantum reader-processors, converged via natural (quantum entanglement) selection, to three measurement operations $J = 3$ in Eq (15) to obtain adequate statistical probabilistic measurements of 20 entangled proton qubit states occupying $G'-5HMC'$ and $*G-5HMC*C$ superposition sites; $*A*-*U$ sites were deleted [16, 38].
The three selected quantum processor measurements identified a triplet code for a precursor tRNA, where L-amino acids were selected. Three separate probabilistic measurement operations would "quantify" enough the 20-different entangled proton qubit states, and therefore, specify about 20, i.e., 22, amino acids for participation in protein structure [57]. The scenario outlined here implies quantum reader measurements of entangled proton qubits occupying ancestral $G'-5HMC', *G-5HMC'$ and $*A*-*U$ superposition sites may have provided the initial quantum informational content, specifying evolutionary parameters for origin of the genetic code, consisting of ~22 L-amino acids specified by $4^3$ triplet codons [35, 36, 37, 38, 39].
EPR-Entanglement Darwinian Polynomial Predictions for Microsatellite Genomic Dynamics
Rationale for Entanglement-Enabled Instability of Microsatellites
Microsatellites of length $L$ are short ($20 \leq L \leq 80$ bp) tandem repeats (STRs) of duplex DNA with repeat unit $\leq 6$ bp [52, 74, 118, 119, 120]. Hundreds of thousands of STRs are distributed throughout eukaryote and prokaryote genomes [112, 119] and have become a primary source of nuclear genetic markers for a variety of applications [120, 122]. Because of considerable variability in repeat number at most loci, i.e., polymorphism, microsatellites are frequently used in the study of evolution and mapping of heritable disease genes [53, 104]. Studies on the origin, evolution and instability of such genes [105, 106, 107, 108, 109, 110, 111] have employed linkage disequilibrium analysis that is dependent on microsatellite mutation rates. Microsatellites generally exhibit mutation by the gain and/or loss of repeat units with an occasional point mutation interruption [104, 109, 110, 111]. Classical explanations for microsatellite evolution are incomplete [38, 123]. STR evolution rates were observed to be different for humans, non-human primates and rodents, implying variable, species dependent mutation rates in STRs [122, 123, 124, 125]. When quantum entanglement algorithm dynamics [15, 16, 17, 37, 38, 54, 104] are neglected, the relative distribution of microsatellites and their individual lengths throughout eukaryotic and prokaryotic genomes were an enigma [52, 74, 112, 118, 119, 120]. The effectiveness of resolving microsatellite evolution data is a function of the accuracy of models for microsatellite evolution [37, 38, 39, 54]. Inadequate models reduce analytical insight and yield misleading conclusions, inconsistent with observation [52, 74, 112]. The development of accurate models, where predictions agree with observations, requires a proper understanding of molecular mechanisms responsible for intra-loci and extra-loci dynamic events [37], and their consequences [38, 39]. In these cases [37, 38, 39] entanglement-enabled molecular mechanisms can cause expansions and/or contractions within microsatellite loci [104, 105], consistent with observation [52, 53, 54]. Evaluation of microsatellite evolution in terms of EPR- generated [29] entangled proton qubits that are “measured by” Grover’s-type [40] quantum processers [36] provides physical and chemical insight into the molecular dynamics responsible for evolutionary distribution of the 22 most abundant microsatellites, STRs, common to rat and human genomes [35, 38, 52]. Time-dependent molecular clock [35, 36, 37, 38, 39, 54, 100, 101, 102] genetic alterations are consistent with Grover’s-type [40] enzyme quantum-readers measuring, δt << 10–13 s, EPR- generated [29, 30, 31] entangled proton qubit states G′-C′, *G- *C, *A-*T (Figure 2-4) to yield time-dependent substitutions [15, 17, 54], ts, and time-dependent deletions [16], td, after quantum information processing, Δtʹ ≤ 10–14 s [13, 35, 36], events of (i) transcription, (ii) translation, (iii) selection of accessible amino acids for peptide bond formation, (iv) random genetic drift [103] and (v)initiation of genome growth. Metastable hydrogen bonding amino (−NH2) protons encounter quantum uncertainty limits, Δx Δpx ≥ ћ/2, which generate probabilities of EPR arrangements, keto-amino ―(entanglement)→ enol−imine, yielding reduced energy entangled proton qubits shared between two indistinguishable sets of electron lone-pairs belonging to decoherence-free subspaces of enol oxygen and imine nitrogen on opposite strands. The evolutionarily selected quantum entanglement algorithm responsible for observable ts and tdhas been operational since the era of ancestral RNA protein genomes [35, 36, 58, 59, 60, 61], thereby providing time- dependent, ‘point’ genetic variation in allsubsequently evolved duplex DNA [100, 101, 102]. Consequently, a time dependent introduction of additional initiation codons UUG, CUG, AUG, GUG and/or stop codons UAA, UAG, UGA can cause the creation of additional polypeptides and/or the absence of “essential” polypeptides, some of which could be responsible for initiation of, or reinitiating, DNA synthesis [37, 38, 39, 104]. Such additional initiating polypeptides could be responsible for adding more repeat units to an original microsatellite. Similarly, “new” termination codons could introduce “truncations” of peptide chains that participate in transcription and/or replication. An accumulation of entangled proton qubits and subsequent transcriptase measurements of entangled qubit states could specify the implementation of initiation codons and deletions or stop codons in microsatellites and/or their flanking sequences [37, 38, 39].
Initiation and Termination Codons via Grover’s measurements of EPR-Generated Entangled Proton Qubits
Observations [15, 16, 17, 20, 21, 22, 52, 53] and analyses [36, 37, 38, 39, 49, 50, 51, 54] imply metastable hydrogen bonding amino (−NH2) protons encounter quantum uncertainty limits, Δx Δpx ≥ ћ/2, which generate probabilities of EPR-created entangled proton qubits [35]. Transcription and replication of entangled proton qubit superposition G′-C′ and *G-*C sites yield observable, time-dependent molecular clock base substitutions, ts [15, 17, 35, 36] G′2 0 2 → T, G′0 0 2 → C, *G0 2 00 → A &*C2 0 22 → T (Table 1) whereas entangled proton qubit states within *A-*T sites, i.e., A-T → *A-*T (Figure 4), exhibit time-dependent deletions, td, *A → deletion and *T → deletion [16]. Also, when G′ and/or *C is located on the transcribed strand, time-dependent substitutions, ts ─ G′2 0 2 → T and/or *C2 0 22 → T are expressed by “Grover’s-type” transcriptase measurements of entangled proton qubits before replication is initiated (Figure 5) [15, 16, 17, 20, 21, 23, 35]. Subsequent replication after entangled enzyme quantum searches, Δt′ ≤ 10−14 s expresses genotypically incorporated ts ─ G′2 0 2 → T and *C2 0 22 → T at frequencies identical to those previously exhibited by quantum transcription before replication [15, 16, 17, 20, 21, 35, 36]. In these cases, G′ → T and *C → T contributions to the “gene pool” are 2-fold > “replication only” expectations [17, 38], caused by transcriptase quantum processing, specifying frequencies of subsequently incorporated ts, G′ → T and *C → T. Based on predictions of quantum entanglement algorithmic processing of EPR-generated entangled proton qubits accumulating with time in metastable duplex DNA base pairs, observed as G-C → G'-C', G-C → *G- C* and A-T → *A-*T [15, 16, 17, 35, 36], the potential for a microsatellite [52, 74] to exhibit expansion or contraction over evolutionary times can be qualitatively specified [36, 104]. This hypothesis based on observation [53, 105, 106, 107] assumes that the evolutionarily selected quantum entanglement algorithm [35] responsible for ts [15, 17, 100, 101, 102, 36] and td [16, 52] has been operational since the era of ancestral RNA protein genomes [58, 59, 60, 61], and therefore, has provided a source of time-dependent, ‘point’ genetic variation in all subsequently evolved duplex DNA [28, 46]. The model also assumes a functional relationship exists between the relative positions of entangled proton qubit states within microsatellites and initiation regions for DNA replication [126]. Consequently, a time-dependent introduction of additional initiation codons UUG, CUG, AUG, GUG could cause the creation of additional polypeptides [35, 36, 104], some of which could be responsible for initiation of, or reinitiating, DNA synthesis [15, 16, 17]. Such additional initiating polypeptides could be responsible for adding more repeat units to an original microsatellite [49, 50, 51, 54]. Similarly, a time-dependent accumulation of stop codons UAA, UAG, UGA could introduce terminations of peptide chains that participate in transcription and replication [16, 35]. Subsequent transcription and resulting DNA synthesis would
| 1 | 2 | 3 | 4 | 5 | 6 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rat Motif | No. bp>60 | Total Rat | Human Motif | No. bp > 40 | Total Human | |||||||||||||
| 1 | CA | 8 | 133 | A | 2 | 131 | ||||||||||||
| 2 | CT | 7 | 56 | CA | 10 | 73 | ||||||||||||
| 3 | A | 0 | 34 | AAAT | 3 | 21 | ||||||||||||
| 4 | AAAT | 0 | 18 | CT | 1 | 19 | ||||||||||||
| 5 | AAGG | 1 | 13 | AT | 3 | 16 | ||||||||||||
| 6 | CAG | 0 | 11 | AAAG | 7 | 15 | ||||||||||||
| 7 | AAAG | 1 | 8 | AAC | 0 | 13 | ||||||||||||
| 8 | AGAT | 1 | 8 | AAAC | 0 | 10 | ||||||||||||
| 9 | AAC | 0 | 7 | CCG | 1 | 10 | ||||||||||||
| 10 | ACGC | 0 | 7 | CAG | 2 | 9 | ||||||||||||
| 11 | AT | 1 | 7 | AGG | 0 | 9 | ||||||||||||
| 12 | AGG | 1 | 6 | AGAT | 7 | 9 | ||||||||||||
| 13 | AATG | 0 | 6 | AAGG | 3 | 8 | ||||||||||||
| 14 | CAGA | 0 | 6 | ATCC | 3 | 7 | ||||||||||||
| 15 | ACC | 0 | 6 | AAT | 2 | 7 | ||||||||||||
| 16 | AAAC | 0 | 5 | AATC | 0 | 3 | ||||||||||||
| 17 | AAT | 0 | 5 | AATC | 0 | 3 | ||||||||||||
| 18 | AGGG | 0 | 5 | ACAT | 0 | 3 | ||||||||||||
| 19 | ACAT | 0 | 4 | ACC | 0 | 3 | ||||||||||||
| 20 | AATT | 0 | 3 | CAGA | 0 | 2 | ||||||||||||
| 21 | ATCC | 0 | 3 | AAG | 1 | 2 | ||||||||||||
| 22 | CCG | 0 | 3 | ATC | 0 | 2 |
consistent with the selected quantum entanglement algorithm for EPR-generated [29, 30, 31] time-dependent molecular clock DNA evolution [15, 16, 17, 100, 101, 102], the model if applicable should predict, qualitatively, the evolutionary distribution of the 22 most abundant microsatellites (Table 3) common to rat and human DNA [38, 52].
Although classical modes of evolution responsible for individual microsatellite length and their relative distribution throughout eukaryotic and prokaryotic genomes have remained an enigma [52, 74, 118, 119, 120, 127, 128], quantum entanglement algorithm processes [35, 36] provide an internally consistent rationale in agreement with observation [38, 52] for relative expansion and/or contraction of specific STRs over evolutionary times [37, 38, 39]. Microsatellite duplexes whose EPR-generated [29, 30, 31] entangled proton qubits are “measured by” Grover’s processors [40] cangenerate a preponderance of initiation codons UUG, AUG, CUG, GUG that participate in the expansion mode of DNA synthesis [37, 49, 50, 54, 105]. But if more termination codons UAA, UGA, UAG were introduced, and/or the sequence consisted exclusively of A-T, such microsatellites would generally decrease in relative abundance over evolutionary times [16, 37, 38]. This model is tested by comparing quantum entanglement algorithm predictions [37] of microsatellite expansion or contraction with observation, for each of the 22 most abundant microsatellites common to human and rat (Table 3). Analyses assumptions are (a) STR evolution is a consequence of EPR-generated [29, 30, 31] entangled proton qubits populating STRs or its flanking sequence that is operated on by Grover’s-type [40] quantum entanglement algorithmic processes [36, 37, 38, 39], which generates molecular clock events, ts and td, and their “dynamic” consequences [16, 104, 105, 106, 107, 108, 109, 110, 111], and (b) the rat genome is more ancient than human [129]. This model is tested by evaluating the evolutionary expansion and contraction “dynamic potential” [37, 104, 105] of the twenty-two most abundant microsatellites (Table 3) common to human and rat [38, 52]. From this list (Table 3), predictions by Grover’s- type [40] quantum information processing of EPR- generated entangled proton qubit states identify two ordered sets – eleven exhibiting expansion and eleven exhibiting contraction – of microsatellites, consistent with observation [35, 38, 52]. Agreement between theory and observation (Tables 4a & 4b) provides an entanglement- based evolutionary rationale for the relative distribution of the 22 most abundant STRs in rat and human genomes [35, 38, 52].
EPR-Enabled Triplet Repeat Expansion and Contraction of Unstable (CCG)n and (CAG)n Microsatellites
Unstable repeat nucleotide sequences are responsible for ~ 20 or so heritable human genetic diseases [106] and have been studied at the molecular level since 1991 [107].
![Figure 10: Base substitution pathways generated by EPR arrangements, _keto-amino → enol-imine_, and introducing entangled proton qubit states in duplex triplet repeats of (a) CCG/GGC and (b) CAG/GTC. The specific substitutions are in parentheses, e.g., (C → T), adjacent to the reactive 5' or 3' strand of the triplet duplex. The initial product is selected by the proton qubit “trapped” in a DNA groove [67-70], δt << 10−13 s, which identifies the participating eigen state of the _G′-C′_ or _*G-*C_ superposition within the triplet duplex. Subsequent transcription (trans) and/or replication (rep) of enol and imine proton qubit isomers within STRs yield altered triplet codes, where pathways for generating initiation codons and stop codons are indicated. The CUG initiation codon can be derived from keto-amino CAG/GTC as indicated. Notation specifying unique proton qubit states is that of Figure 5.)](/fulltextimages/1462/fig_10.png)
Figure 10a: Entangled proton qubits populating CCG repeats. (Figure 10: Base substitution pathways generated by EPR arrangements, keto-amino → enol-imine, and introducing entangled proton qubit states in duplex triplet repeats of (a) CCG/GGC and (b) CAG/GTC. The specific substitutions are in parentheses, e.g., (C → T), adjacent to the reactive 5' or 3' strand of the triplet duplex. The initial product is selected by the proton qubit “trapped” in a DNA groove [67, 68, 69, 70], δt << 10−13 s, which identifies the participating eigen state of the G′-C′ or *G-*C superposition within the triplet duplex. Subsequent transcription (trans) and/or replication (rep) of enol and imine proton qubit isomers within STRs yield altered triplet codes, where pathways for generating initiation codons and stop codons are indicated. The CUG initiation codon can be derived from keto-amino CAG/GTC as indicated. Notation specifying unique proton qubit states is that of Figure 5.)
However, internally consistent classical mechanisms responsible for microsatellite repeat, intergenerational instabilities [53, 105, 106, 107, 108, 109] and subsequent expressions of diseases have been an enigma [110, 111]. Insight into microsatellite instabilities is implied by consequences of the quantum entanglement algorithm operating on, for example [35], (CCG)n and (CAG)n microsatellites [52, 104] listed in Table 3. In cases of fragile X syndrome (FX), triplet repeats, (CCG)n, are in the 5'-UTR of gene FMR1 where data indicate a maternal bias and a high upper limit expansion copy number of ~ 2000 (CCG)n repeats [104, 107]. Figure 10a illustrates that entangled proton qubit states in (CCG)n repeats would not generate stop codons, but three of the twelve ts pathways (25%) could introduce entangled proton qubit states that could be measured to express an initiation codon, 5′-CUG-3′. Specifically, entangled proton qubits could accumulate for years to decades in oocyte DNA [130] before the enzyme quantum reader would “measure” 5'-C*C2022G-3' and 5'- CG'202G-3' entangled qubits, thereby expressing CUG via transcription of entangled proton qubit states (Figure 5) and subsequent replication. Since rates of accumulating entangled proton qubit states in haploid DNA would be smaller in ~34 0C sperm [131] than in 37 0C oocyte genomes [28, 35, 100], hyperexpansions (copy no. >1000) of (CCG)n in oocyte DNA versus limited expansions (copy no. < 1000)in malehaploid DNA [104, 107] are attributed to an increased energy density of duplex DNA [132, 133] in 37 0C oocyte genomes [35, 37, 100]. At transcription before replication, δt ≤ 10–13 s, accumulated entangled proton qubit states, 5'-C_*C202_2_G- 3' and 5'-C_G'202_G-3', would express additional “new” initiation codons, 5'-CUG-3'. In the “neighborhood” of initiation regions for DNA replication [126], such additional reinitiating signals could cause an addition of more triplet repeats, which would be manifested as (CCG)n expansion [104, 105, 106, 107, 108]. Thus, at transcription just before fertilization of an oocyte [130], accumulated entangled proton qubit superposition states C*C202_2_G and C_G'202_G (Tables 1 & 2, Figure 10a) could be transcribed to yield CUG, which can also specify reinitiating of DNA synthesis, thereby causing massive (CCG)n expansion in oocyte DNA [104, 105, 106, 107, 108]. Figure 10a also shows that twelve of the twenty-one (57%) _ts that code for an amino acid would result in amino acid substitution.
Figure 10b: Entangled proton qubits populating CAG repeats. Unstable CAG repeats are responsible for several neurological diseases [35, 105, 106], including Huntington’s disease [53, 54, 109]. Figure 10b illustrates that EPR-generated entangled proton qubits accumulated in CAG-repeats could express initiation codons, AUG and GUG, which would require replication, whereas the two UUG codons could be expressed by quantum transcription prior to replication [35, 36, 37]. Similarly, the two UAG stop codons would be expressed by quantum transcription before replication. Observation that CAG expansion is more efficient in sperm [134, 135, 136] implies that replication-dependent pathways would be primarily responsible for CAG expansion in haploid human DNA. Expression of UAG codons would be responsible for nonsense mutations, and thus, contractions are observed in CAG tracts from sperm [136]. This combination of expansion and contraction modes would govern instability exhibited by CAG repeats [37, 53, 54, 136]. Figure 10b illustrates that 5'-CAG-3' and 5'-CTG-3' are complementary components on opposite strands of a duplex repeat, 5'-CAG/GTC-5'. However, ordinary keto- amino 5'-CTG-3' could generate the CUG initiation codon by transcription without entanglement-enabled ts intervention. Amino acid substitutions would be introduced in four of the ten ts that code for amino acids. Specifically, Gln would be replaced by Glu, Lys and His (twice).
EPR-Enabled Expansion and Contraction of Dinucleotide Repeats
Expansion and contraction evolutionary dynamics of CA and CT repeats (Table 3)are compatible with the quantum entanglement algorithm, triplet repeat expansion mechanism [37, 38, 54, 104] where dinucleotide repeats of (CA)3n and (CT)3m can be treated as hexarepeat sequences of alternating triplets, i.e., [(CAC)(ACA)]n and [(CTC)(TCT)]m. Accordingly, expectations are considered for consequences of EPR- generated entangled proton qubits populating metastable G C and A-T sites in these triplet components of hexarepeat sequences. The eight pathways by which entangled proton qubits populate each of the two triplet duplexes, CAC/GTG and CTC/GAG, are illustrated in Figures 11a-b. Note that the keto amino 5' GTG 3' component in Figure 11a could yield an initiation code, or could specify valine. Additionally, four of the eight pathways for introducing entangled proton qubit states into the CAC/GTG duplex could generate an initiation codon, all of which would be derived from GTG strands. Since ts stop codes are absent, and initiation codons constitute 50% of the possible yield generated by introducing EPR-entangled proton qubits into the CAC/GTG duplex (Figure 11a), the model predicts that hexarepeat duplexes of (CACACA/GTGTGT) would generate high levels of the expansion mode of DNA synthesis. This would add more CA repeats to the original sequence. Transcription of entangled proton qubit states – and subsequent replication of the corresponding decohered isomers [37, 38, 39] – occupying A T rich triplet duplexes of ACA/TGT and TCT/AGA would yield only amino acid substitutions and deletions at *A-*T. (Data on A-T rich triplets are not displayed). Analogous pathways for introducing entangled proton qubit states within the CTC/GAG duplex (Figure 11b) indicate that probabilities are approximately equal for generating the initiation codon, CUG (G'0 0 2 →C), and the stop codon, UAG (G'2 0 2 →T). However, CUG codons require replication for expression, whereas UAG codons can be expressed by transcription before replication. The higher probability, 50%, for superposition entangled proton states in CAC/GTG to yield an initiation code, versus that for CTC/GAG, 12.5%, implies a greater

Figure 11a: Entangled proton qubits populating CAC/GTG triplets.
Figure 11b: Entangled proton qubits populating CTC/GAG triplets.
(Figure 11 Pathways for generating entangled proton qubit states in STRs of (a) CAC/GTG and (b) CTC/GAG where decohered isomers are replicated to yield time- dependent substitutions, ts. The specific substitutions are in parentheses adjacent to the reactive 5′ or 3′ strand of the duplex triplet. The initial product identifies the “selected” eigenstate of the G′-C′ or *G-*C quantum superposition within the STR, using Figure 5 notation. Subsequent transcription (trans) and/or replication (rep) of STRs yield the resulting triplet codes. Pathways for entangled proton qubit states to generate initiation and stop codons are indicated. Introduction of the 5′-GUG-3′ initiation codon by the keto-amino CAC/GTG duplex is shown. Notation specifying states of entangled proton qubits is given in Figure 5 & Table 2) probability for expansion by CA repeats compared to CT. Also, the terminationcode 5′−UAG−3′, illustrated in Figure 11b is absent from entangled proton qubit states occupying CAC/GTG duplexes (Figure 11a). Repeat sequences (CA)n should therefore be more numerous and longer than (CT)min both rat and human, consistent with Table 3, Figure 11a-b and observation [37, 38, 52, 74, 118, 119].
EPR-Generated Instabilities in Microsatellites Common to Rat and Human Genomes
Beckmann and Weber [52] observed that 43% of rat microsatellites are of length > 40 bp compared to only 12% of human repeat sequences. This agrees with Love et al. [119] who found that most mouse microsatellites are longer than corresponding sequences in the human genome. Data on STRs in Table 3 show that 11 of the 22 STRs are more abundant in ancient rat than human (Table 4a), and thus, 11 of the 22 STRs are less abundant in rat than in human homologues (Table 4b). Compared to human DNA, the percentages increase and decrease in relative abundance of STRs in the rat genome are given in column 5 of Tables 4a and 4b, respectively. For example, in the case of CT (Table 4a), the 195% increase in relative abundance is given by [(56 – 19)/19] × 100.
| 1 | 2 3 | 4 | 5 | 6 | 7 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Motif | Relative Abundance | #H/#R | % Increase Relative | % Pathways | % Pathways STOP | ||||||||||||
| (Number of STRs) | Abundance | INITIATION Codons | Codons | ||||||||||||||
| Rat | Human | ||||||||||||||||
| ACGC | 7 | 1 | 0.14 | 600% | 33% | 8.30% | |||||||||||
| AGGG | 5 | 1 | 0.2 | 400% | 16.60% | 8.30% | |||||||||||
| CAGA | 6 | 2 | 0.33 | 200% | (a) 50% | 25% | |||||||||||
| CT | 56 | 19 | 0.34 | 195% | 12.50% | 12.50% | |||||||||||
| (b)ACC | 6 | 3 | 0.5 | 100% | 32% | 25% | |||||||||||
| CA | 133 | 73 | 0.55 | 82% | 50% | 0% | |||||||||||
| AAGG | 13 | 8 | 0.62 | 63% | 25% | 25% | |||||||||||
| CAG | 11 | 9 | 0.82 | 22% | 50% | 25% | |||||||||||
| AATG | 6 | 3 | 0.5 | 100% | (c) 0% | 75% | |||||||||||
| ACAT | 4 | 3 | 0.75 | 33% | (d) 0% | 50% | |||||||||||
| AATT | 3 | 1 | 0.33 | 200% | 0% | (e) 0% |
| 1 | 2 3 | 4 | 5 | 6 | 7 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Motif | Relative Abundance | #H/#R | % Decrease Relative | % Pathways | % Pathways | |||||||||||||
| (Number of STRs) | Abundance | STOP Codons | INITIATION Codons | |||||||||||||||
| Rat | Human | |||||||||||||||||
| A | 34 | 131 | 3.85 | 74% | 0% | 0% | ||||||||||||
| AT | 7 | 16 | 2.29 | 56% | 0% | 0% | ||||||||||||
| AAT | 5 | 7 | 1.4 | 29% | (a)0 % | 0% | ||||||||||||
| AAAT | 18 | 21 | 1.17 | 14% | (a)0 % | 0% |
| ATCC | 3 | 7 | 2.33 | 57% | (b)75% | 25% |
|---|---|---|---|---|---|---|
| AAAC | 5 | 10 | 2 | 50% | 50% | (c)0 % |
| AAAG | 8 | 15 | 1.88 | 47% | 25% | 25% |
| AAC | 7 | 13 | 1.86 | 46% | 50% | (d)0 % |
| AGG | 6 | 9 | 1.5 | 33% | 25% | 25% |
| AGAT | 8 | 9 | 1.13 | 11% | (e)50% | 25% |
| CCG | 3 | 10 | 3.33 | 70% | 0.00% | 25% |
in columns 6 & 7, respectively. These reaction results are illustrated in Figure13a, f. With exception of CT (CTC, Figure 11b) and AAGG (Figure 12d), whose entangled proton qubit states would be equally likely to generate initiation codons and stop codons, six of the initial eight motifs listed in Table 4a satisfy the criteria for the expansion mode of DNA synthesis. This is represented inTable 4a, columns 6 & 7, where, for the specific STR, the probabilities for entangled proton qubit states to generate initiation codons are greater than those for introducing stop codons. Using these initial eight motifs, i.e., ACGC through CAG, average values for columns 6 & 7 are 34% for initiation codons and 16% for stop codons. These STRs would thus be expected to exhibit the expansion mode of DNA synthesis, and consequently over evolutionary times, increase their abundance in ancient genomes. This is consistent with the 600% to 22% increases in relative abundance of these eight STRs in the rat genome, listed in column 5 of Table 4a, where the average increase is 208%. The percentages increase in relative abundance of 600%, 400% and 200% exhibited by ACGC, AGGG and CAGC, respectively, can be attributed, in part, to the transcriptionally enhanced “base dose” effect caused by enzyme quantum reader measurements of entangled proton qubits generating quantum transcription expressions of substitutions, *C2 0 22→T and G'2 0 2 →T, before replication, and also, expressed after T22 0 22 is genotypically incorporated by replication [15, 16, 17, 35, 38]. Consequently, initiation codons AUG, GUG and UUG illustrated in Figure 12a-c could be expressed by transcription before replication and likewise after replication, thereby generating a 2-fold enhanced “base dose” effect for these initiation codons. In the case of ACGC (Figure 12a), the four pathways for introducing initiation codons (AUG, GUG) can be expressed by transcription before replication, but introduction of UGA requires replication before expression is allowed. These more numerous pathways for expressing AUG and GUG by transcription imply an advantageous increase in relative abundance of ACGC, consistent with Table 4a. Figure 12b illustrates how transcriptase quantum processing of G'2 0 2 in STRs of AG'202GG and AGG'202G can yield 5'-AUG-3', 5'-GUG-3' and 5'-UGA-3'. In these situations, pathways for initiation codons are 2-fold greater than those for stop codons. Thus, one should expect AGGG to exhibit an increase in relative abundance, consistent with Table 4a. Figure 12c shows that AUG and GUG require replication for expression, whereas UUG and UAG are equally likely to be expressed by transcriptase quantum processing of *C2 0 22 within a CAGA/GTCT duplex.

Figure 12a: ACGC: EPR-generated dynamic mutations. (Figure 12 Pathways for entangled proton qubit introduction ofdynamic mutations expressed as initiation codons and stop codons in STRs of (a) ACGC, (b) AGGG, (c) CAGA, (d) AAGG, (e) AATG and (f) ACAT. Pathways for expressing initiation and stop codons by keto-amino STRs are indicated.)
Figure 12b: AGGG: EPR-generated dynamic mutations.
Figure 12c: CAGA: EPR-generated dynamic mutations.
![Figure 14: (CAG)n repeat length versus age-of-onset of Huntington’s disease (adapted from Figure 3 of Gusella et al. [53]).](/fulltextimages/1462/fig_14.jpeg)
Figure 12d: AAGG: EPR-generated dynamic mutations.
Figure 12e: AATG: EPR-generated dynamic mutations. Additionally, the 5'-CUG-3'codon can be expressed by transcription of the keto-amino state from the GTCT strand. These options imply an increase in relative abundance of CAGA, as observed. Table 4a identifies keto- amino states of AATG/TTAC (Figure 12e) and ACAT/TGTA (Figure 12f) that could allow the AUG initiation codon to be expressed. Such availability of an initiation code derived from the keto-amino state of a STR implies additional contributions to the expansion mode of DNA synthesis, without introducing entangled proton qubit states. This provides a rationale for the more abundant AATG and ACAT in rat DNA (Table 4a). Unlike AATT in Table 4a, which shows a 200% increase in relative abundance in rat, the four-other exclusive A-T motifs in Table 4b A, AT, AAT, AAAT exhibit decreases in relative abundance, yielding an average of − 44% (column 5) which is consistent with model expectation. The greater abundance of AATT in the more ancient genome (Table 4a) implies that dominant evolutionary pressures on AATT are different than those for the other pure A-T motifs.As illustrated in Figure 13e,
Figure 12f: ACAT: EPR-generated dynamic mutations. Sequences of AATT could be derived from a G'2 0 2 → T substitution in AATG. This would allow keto-amino AATG to express the initiation codon, 5'-AUG-3', which could then operate on downstream sequences of AATT, resulting in expansion of the latter. The 200% relativeincrease in sequences of AATT could be derived from a G'2 0 2 → T substitution in AATG. This would allow keto-amino AATG to express the initiation codon, 5'-AUG- 3', which could then operate on downstream sequences of AATT, resulting in expansion of the latter. The 200% increase in relative abundance implies that the availability of (keto-amino) AATG to express 5'-AUG-3' causes the expansion mode to be dominant over td in AATT sequences. The fact that AATG exhibits only 100% increase in relative abundance (compared to 200% for AATT) is consistent with Figure12e, which illustrates entangled proton qubit states in AATG/TTAC duplexes yield stop codons. These time-dependent truncation pathways are not available to AATT, but keto-amino AATT allows UAA expression. Evidently in this case, time- dependent stop codes generated by AATG exert a dominant influence; so, compared to AATG, downstream AATT sequences could yield greater relative abundance in ancient genomes, consistent with observation. The remaining sixmotifs containing G-C in Table 4b (excluding CCG, Figure 10a), i.e., ATCC through AGAT, also show decreases in relative abundance where, compared to human DNA, the average decrease is − 41%. However, entangled proton qubit states populating the STRs of AAAG (Figure 13c) and AGG (Figure 13e)show equal probabilities for generating stop and initiation codons, whereas the four other STRs exhibit greater probabilities for generating stop codons from transcriptase measurement ofentangled proton qubit states (column 6) in Table 4b. The average percentages for introducing stop codons and initiation codons are, respectively, 48% and 17% for these six motifs in Table 4b. This implies reductions in relative abundance as a function of increased entangled proton qubit states accumulated within these STRs. Figure 13a shows that the eight pathways for introducing entangled proton qubit superpositions into the ATCC/TAGG duplex could yield seven stop codons and two initiation codons. This introduction of additional stop codons exerts the dominant influence on the evolution of ATCC microsatellites, consistent with the observation that ATCC is 21st in the more ancient rat genome and is 14th in human DNA. Both AAAC (Figure 13b) and AAAG (Figure 13c) repeats are A-T rich and are therefore susceptible to deletions of *A-*T sites. This would yield decreases in abundance as a function of increased EPR-generated accumulation of entangled proton qubit states, illustrated in Table 4b. In the case of AAAC (Figure 13b), two of the four EPR-dependent pathways generate UAA because of transcriptase quantum processing of AAA_*C202_2, but the 5'-UUG-3' initiation codon is available from the keto- amino state of TTTG.Long-term accumulation of entangled proton qubit states in AAAC/TTTG duplexes introduces stop codons, UAA, by 50% of the EPR-enabled pathways (Table 4b), and the original keto-amino initiation code, TTG, would be removed, thereby diminishing the relative abundance of AAAC microsatellites in ancient DNA. In the case of AAAG (Figure 13c), one pathway (25%) can generate the stop codon, UAA, by transcription before replication, and the initiation codon, UUG, is introduced after replication by one pathway (25%). Thus, compared to AAAG (7th in rat), one could expect AAAC (16th in rat) repeats to be less abundant in the more ancient rat genome, consistent with observation and Table 4b. However, AAAC exhibits a decrease in relative abundance of −50% and AAAG shows a decrease of −47% in relative abundance (Table 4b). Similarly, long term accumulation of entangled proton qubit states in AAC/TTG duplexes (Figure 13d) would replace the original keto-amino initiation codon, UUG, with UAA codons. This would cause AAC to become less abundant in rat (15th) compared to human (7th).Although AGG (Figure 13e) exhibits equal probabilities for generating stop and initiation codons, the A-T rich AAC(Figure 13d)shows a slightly greater decrease in relative abundance, i.e., −46% versus −33% for AGG (Table 4b). Note also that the relatively small difference in abundance of AGG in rat (12th) and human (11th) DNA (Table 3) is compatible with approximately equal probabilities of generating stop and initiation codons where the A-T site would exhibit deletions.
![Figure 15: Cancer incidence as a function of age (ages 10 to 80y) [56] (Average age distribution of all “Class 1” tumors (those with single incidence peak at age > 50 y) classified by the Connecticut Tumor Registry between 1968 and 1972). In this case, EPR-generated, quantum entanglement $$](/fulltextimages/1462/fig_15.png)
Figure 13a: ATCC: EPR-generated dynamic mutations. (Figure 13 Pathways for entangled proton qubit introduction ofdynamic mutations expressed as initiation codons and stop codons in STRs of (a) ATCC, (b) AAAC, (c) AAAG, (d) AAC, (e) AGG and (f) AGAT. Pathways for expressing initiation and stop codons by keto-amino STRs are indicated.) The decrease in CCG relative abundance (Figure 10a) in the more ancient rat genome is consistent with G-C replacement by A-T or T-A in three of the four pathways generated by the quantum entanglement algorithm operating on (CCG)n repeats [35, 37, 38, 54]. Qualitative agreement between data in Tables 4a & 4b and model expectation also includes the observation that 8 of the 11 STRs, which are less abundant in rat (Table 4b), are A-T rich compared to only 3 of the 11 more abundant STRs in Table 4a.
![Figure 16: Alzheimer’s disease incidence as a function of age. (LOAD (late onset Alzheimer’s disease) affects approximately 2% of the age 65 y population, and doubles approximately every 5 y thereafter, yielding incidence > 50% at age 90 y [77].)](/fulltextimages/1462/fig_16.jpeg)
Figure 13b: AAAC: EPR-generated dynamic mutations.
Figure 13 c: AAAG: EPR-generated dynamic mutations.
Figure 13d: ACC: EPR-generated dynamic mutations.
Figure 13e: AGG: EPR-generated dynamic mutations.
![Figure 17: This model displayed in Figure 17 implies life’s origin emerged, and was sustained, in terms of approximate evolutionary increments [35-39]. A set of increment arrows (→) in Figure 17 identifies the following sequential (or simultaneous) evolutionary developments over the three phases of primordial evolution**:** monomers → oligomers → ribozymes → duplication of nucleotides → duplex RNA polymer segments → entangled proton qubits → ribozyme – proton entanglements → quantum transcription of entangled qubit states → quantum translation of entangled qubit message → quantum selection of triplet code → construct polypeptides → enzymes from ribozyme – proton entanglements → replication via enzymes → introduction of repair enzymes → genome chemistry selection, RNA replaced by DNA → duplex DNA genomes, etc. This OoSL model implies quantum entanglement algorithms, developed and implemented in ancestral RNA – protein systems, were subsequently retained and refined within evolving duplex DNA systems. Classical mechanisms do not explain evolutionary processes between ~ –4.1×109 y and ~ – 3.7×109 y [57,63,83], but EPR-entanglement algorithm processes [7-11] provide plausible mechanistic explanations [35-39]. This argument allows explanations for origins of quantum information processing algorithms exhibited by ancient [46] T4 phage DNA [15-17], rodent – human microsatellite DNA evolution [38,52], and 21st- century human genomic systems [37,39,47- 51,54,100,104]. In this case, a reverse-time extrapolation — of proton quantum dynamics required for observable quantum information processing exhibited by ancient [46] T4 phage DNA [15-17] — appears applicable to analogous, metabolically inert [64], duplex RNA segments occupying primordial pools (0 0C to 20 0C, pH 7 [18- 22,36,89]) of ribozyme ‒ RNA systems [57-61].](/fulltextimages/1462/fig_17.png)
Figure 13f: AGAT: EPR-generated dynamic mutations.
Quantum ProcessingImplications of EPR- Generated Entangled Proton Genome Qubits
Confidence in entanglement-enabled bio-molecular information processing [35, 36, 40] is provided by the fact that multiple lines of experimental observation prokaryotic T4 phage systems [15, 16, 17] and human gene systems [37, 39, 49, 50, 51, 54, 104] converge with the EPR- generated, time-dependent molecular evolution model for human – rat STRs herein analyzed and discussed [38, 52]. Metastable amino (–NH2) hydrogen bonded protons are subjected to quantum uncertainty limits, ΔxΔpx ≥ ħ/2, which introduces probabilities of EPR-generated entangled proton qubit superposition states, keto-amino ―(entanglement)→ enol−imine, in STRs. Grover’s [40] quantum processor measurements, δt << 10–13 s, of dynamic entangled proton qubit states predict a time- dependent creation of initiation codons, stop codons and deletions [15, 16, 17, 49, 50, 51, 54], and consequently, provide a mechanism (a) for stochastic random genetic drift, ts + td [100, 101, 102, 103], and (b) for expansion and contraction of STRs [35, 37, 38, 39, 104, 105, 106, 107, 108, 109, 110, 111]. In addition to quantum chemical analyses identifying two internally consistent, “ordered sets” of expanding and contracting STRs from the list of twenty-two (Table 3) most abundant STRs common to human and rat [52], Table 4a provides molecular insight into dynamic mechanisms responsible for evolutionary “expansion” processes. For example, a rationale is presented for (CA)n repeats to be longer and more numerous than (CT)m repeats in both human and rat, consistent with observation [52, 74]. General agreement between model expectation and observed relative abundance of STRs in rat and human genomes [38, 52] implies that evolutionary processes of expansion and/or contraction [104, 105, 106, 107, 108, 109, 110, 111] can be simulated in terms of EPR- generated [29, 30, 31] entangled proton qubit superposition states populating G'-C', *G-*C&*A-*T sites in STRs [15, 16, 17, 123]. These entangled proton qubit statesare subsequently processed by Grover’s quantum processors [35, 36, 37, 38, 39, 40]. An apparent unrecognized consequence of quantum information processing [35, 36, 37, 38, 39, 40] of EPR- generated [29, 30, 31] entangled proton qubits [15, 16, 17, 23] includes genetic instabilities of STRs [104, 105, 106, 107, 108, 109, 110, 111, 123] and phenotypic expression as a function of time (age) of associated triplet repeat human diseases [37, 38, 39, 53, 54, 104, 105, 106, 107, 108, 109, 110, 111], including ALS [78, 79]. The list of 22 microsatellites in Table 3 identifies two “ordered sets” of expanding and contracting STRs shown in Tables 4a-4b. Inspection of Table 3 allows the creation of Tables 4a & 4b, columns 1 through 5. However, percentages of initiation and stop codons listed in columns 6 & 7 are obtained from consequences of metastable hydrogen bonding amino (−NH2) protons encountering quantum uncertainty limits, Δx Δpx ≥ ћ/2, which generates probabilities of EPR arrangements, keto- amino → enol-imine, where position – momentum quantum entanglement [7, 8, 9, 10] is introduced between separating enol and imine protons [35, 36, 37, 38, 39]. Reduced energy product protons are each shared between two indistinguishable sets of electron lone-pairs belonging to enol oxygen and imine nitrogen on opposite strands (Figure 1-4), and consequently, participate in entangled quantum oscillation at ~ 4×1013 s−1 (Tables 8-9, Appendix II) between near symmetric energy wells in decoherence- free subspaces [11, 67, 68, 69] until “measured by” [41, 42, 43, 44, 45] a “Grover’s-type” [40] enzyme quantum processor [35, 36, 37, 38, 39]. Values in columns 6 & 7 (Tables 4a-b) provide explanations for the percentages increase and decrease in relative abundance column 5 in Tables 4a & 4b, respectively. Quantum entanglement algorithm expectations [37, 38, 39] identify a rationale for two ordered sets of evolving microsatellites 11 exhibiting expansion (Table 4a) and 11 exhibiting contraction (Table 4b) consistent with observable relative abundance [52]. Results of subsequent quantum entanglement algorithmic processing of entangled proton qubits are illustrated in Figure 10-13. This agreement between quantum entanglement algorithm predictions [35, 37, 38] and observation [52] provides an internally consistent, microphysical model for the distributions of the 22 most abundant rat and human microsatellites [52, 74, 118, 119, 120]. This quantum information processing model for genomic evolution [35, 36] is in terms of Grover’s [40] processors measuring, and implementing, quantum informational content embedded within EPR-generated entangled proton qubits [37, 38, 39], which cannot be simulated by classical models [52].
Quantum entanglement algorithm analyses [35, 37, 38, 39]of (a) evolving distributions of human-rodent STRs [52, 74, 118, 119, 120], (b) ancient T4 phage DNA [15, 16, 17, 20, 21, 22, 23], and (c) human gene systems [49, 50, 51, 54] are consistent with the hypothesis that ancestral RNA – ribozyme duplex segment systems initially acquired and “processed” EPR- generated entangled proton qubits [7, 8, 9, 10], before the last universal cellular ancestor (LUCA) [57, 58, 59, 60, 61]. Subsequent selection of enzyme proton entanglement processing of entangled proton qubits was an “early” adaptive mutation [28, 57, 137] that allowed development and growth of an increasingly complex, evolving and dynamic [104, 105] genomic system. The fact that EPR-generated ts and td can introduce and eliminate initiation codons UUG, CUG, AUG, GUG and termination codons UAA, UGA, UAG implies resultant “dynamic” mutations [35, 37, 38, 39, 53, 54, 104, 105, 106, 107, 108, 109, 110, 111] played significant roles in physical genomic growth, which has provided the classical duplex molecular matrix on which the quantum entanglement algorithm operates on dynamic, EPR-generated entangled proton qubits [15, 16, 17, 35, 36]. Availability of enzyme – proton entanglement processing [40] of entangled proton qubits [15, 16, 17, 35] allowed growth in genomic mass, i.e., additional base pair units via “expansion” [37, 38, 54, 104, 105, 111], which enhanced the probability introducing additional EPR- generated entangled proton qubits. Most of life’s evolutionary stages e.g., precellular [36, 57, 58, 59, 60, 61], cellular [28, 100, 101, 102, 103], eukaryogenesis [57, 138], etc. have successfully emerged under conditions of continuous accumulations of entangled proton qubits deciphered by quantum processinginformation enzymes (QPIE) to yield ts and td [35], but several evolutionary consequences have not been “accurately” recognized [37, 38, 39, 54]. For example, the quantum entanglement evolution model [35, 36, 37, 38, 39] predicts that “delayed” phenotypic manifestation of Huntington’s disease [53] is a consequence of time required for EPR-entangled proton qubits to populate the “threshold limit” of the conserved huntingtin gene, after which Grover’s [40] processors measure quantum informational content to exhibit phenotypic expression [37, 38, 39, 54]. Based on the evolution scenario outlined here, over the past ~ 3.6 or so billion y [35, 36, 57, 58, 59, 60, 61], enzyme quantum- reader processing [39, 40] of EPR-generated entangled proton qubits has provided an entanglement resource for quantum dynamical genomic growth and evolution, from relatively primitive pre-LUCA systems [35, 36, 58, 59, 60, 61] into the more complex and biologically diverse, modern mammalian genomic system [100, 101, 102, 138]. This growth and development in operational biological complexity is thus a consequence of Darwinian selection operating on enzyme – proton entanglement processes, driven by availability of EPR-generated entangled proton qubits at the microphysical, entangled proton qubit genomic level. This and other reports [35, 36, 37, 38, 39] argue that the smallest enzymatically measurable unit of genetic information is an “entangled pair” of EPR-generated [29, 30, 31] proton qubits, occupying decoherence-free subspaces [11, 67, 68, 69], and subsequently measured by Grover’s [40] quantum processor. Consequently, a nucleotide is not the smallest “basic” unit of genetic information measured by enzyme processors, responsible for communicating time- dependent [100] molecular genomic evolution [36, 37, 38, 39]. In duplex DNA of human genomes, unstable repeats [52, 53, 104, 105, 106, 107, 108, 109, 110, 111] exhibit expansions and contractions via dynamic mutations [35, 37, 38, 39, 54, 104, 105], where (CAG)n sequences (n > 36) can exhibit expansions ≥ 10 (CAG) repeats in 20 y [35, 53, 109]. This observation implies the hypothesis that susceptible ancestral genomes implemented dynamic mutation expansions as consequences of specific ts + td [15, 16, 17, 37, 38]. A “net” triplet repeat dynamic mutation expansion rate of 13 repeats, e.g., (CAG)13 = 39 bp, per 20 y for 3.5 billion y would generate a genome of ~ 6.8×109 bp, which is “ballpark” compatible with bp content of the Homo sapiens’ genome [28, 57]. Based on the present and other [35, 36, 37, 38, 39] assessments, evolutionary genomic growth was, and is, a consequence of the quantum entanglement algorithm introducing, and eliminating, initiation codons UUG, CUG, AUG, GUG and stop codons, UAA, UAG & UGA. This hypothesis is consistent with the fact that overall microsatellite content in a genome correlates with genome size of the prokaryote or eukaryote organism [112]. Selected “expansion” sequences were exploited as conserved genes, e.g. [53, 55, 75, 76, 77, 78, 79], whereas “other” expansion sequences have been relegated to “unspecified” conserved noncoding genomic space (CNGS) [113, 114]. An “accurate” understanding of quantum entanglement algorithm evolution of STRs provides new and useful insight into unusual behavior exhibited by Huntington’s disease (CAG)n repeats [35, 37, 53, 54, 109] and other unstable triplet repeat diseases [78, 79, 104, 105, 106, 107, 108, 109, 110, 111]. Specifically, quantum entanglement algorithm analyses of STR evolution data Tables 3, 4a, 4b support the hypothesis that the ~ 2 to~ 12 yr. delay, after birth, in expression of Huntington’s disease by an inherited long, (CAG)70 repeat [53], is due to the necessity of Grover’s [40] transcriptase quantum processors measuring available entangled proton qubits occupying a “threshold limit” [37] of the inherited (CAG)n (n ≥ 70) repeat [35]. The quantum entanglement algorithm [35, 36, 37, 38, 39] generates a probabilistic yield of entangled proton qubit states, which would manifest an irregular ‘tick rate’, as observed [100, 101, 102, 103]. Also, the expression of mutagenic codes, i.e., expansions and/or contractions, would introduce additional variations into microsatellite molecular clock data [54, 104, 123]. Thus, the quantum entanglement algorithm provides a plausible mechanism at the microscopic entangled proton qubit information level [35, 36, 37, 38, 39] for generating differences in substitution [15, 17], ts, and deletion [16], td, rates. By incorporating these measurable features [15, 16, 17, 35, 36, 37, 38, 39] into models where mathematical variables and operations represent quantifiable biological reality, one could aim for a reduction in parameters and an improved accuracy in models that analyze genetic distance between species [100, 101, 102, 103, 129, 138].
Based on the high level of qualitative agreement between model prediction and observation of STRs evolutionary distributions [52], this analysis concludes that microsatellite evolution [53, 54, 104, 105, 106, 107, 108, 109, 110, 111] can be simulated in terms of EPR-generated, entangled proton qubit states measured by Grover’s [40] quantum processors which introduce ts + td that can cause expansions and contractions [37, 38, 39]. Agreement between model predictions [37, 38] and observed STR evolution [52] of the 22 “most abundant” microsatellites (Table 3) implies significant elements of “correctness” regarding EPR-generated, molecular mechanisms responsible for genome and microsatellite evolution, and thus, warrants further theoretical and experimental investigations.
Polynomial Development
To elucidate factors responsible for age-related disease expression, as a function of acquiring SNPs [15, 16, 17, 18, 19, 20, 21, 22, 23, 35, 36, 37, 38, 39, 49, 50, 51, 54, 55, 56, 75, 76, 77, 78, 79] ts + td plus classical ‘point’ mutations [28, 71, 139] a Darwinian polynomial is constructed that includes both classical [28, 71, 139] and EPR-entanglement [35, 36, 37, 38, 39] contributions. Here manifestations of three different SNP-sensitive age-related diseases cancer [55, 56, 75, 140, 141, 142, 143], Alzheimer’s disease [76, 77, 144, 145, 146], Huntington’s disease [37, 53, 54, 109, 110] are analyzed in terms of classical and EPR-entanglement contributions by a Darwinian polynomial [35, 37, 38, 39, 49, 50]. Most discussions of biological noise, N(t) [101, 102, 103, 147, 148, 149, 150], are in terms of Muller’s [71] classical, constant “mutational load” model, dN/dt = $\lambda$, which neglects EPR-entanglement contributions [35, 36, 37, 38, 39]. Since quantum entanglement terms [7, 8, 9, 10, 11, 35, 36, 37, 38, 39] cannot be efficiently simulated by classical mechanisms [11, 12, 43, 44, 45], the time derivative of total biological noise can more accurately be expressed in terms of an exclusively classical component, $\lambda$ [71], plus quantum entanglement contributions, $\beta t$ [35, 36, 37, 38, 39]. A 3-level quantum approximation for the probability, $P_p(t)$, of EPR-arrangement, $keto-amino \rightarrow enol-imine$, is given by $P_p(t) = \frac{1}{2}(\gamma_p / \hbar)^2 t^2$ (See Appendix I & [49, 50]), where $\gamma_p$ is the energy shift between the initial metastable and final product entanglement state, and $\rho = 1, 2$ for symmetric, asymmetric channels (Figure 2-3), and $h$ is Planck’s constant divided by $2\pi$. Thus dP/dt = $(\gamma_p / \hbar)^2 t = \beta t$, where $\beta = (\gamma_p / \hbar)^2 \approx 2 \times 10^{-23} s^{-2}$ (Cooper, unpublished results); so, the time derivative of the total biological noise, dN/dt, accumulating in the particular gene, $g$, is more accurately expressed by the sum of classical [28, 71] plus EPR-entanglement contributions [35, 36, 37, 38, 39], given as
$$\frac{dN}{dt} = \lambda + \beta t$$
Quantum entanglement contributions are approximated by the $\beta t$ term in Eq (17), which is purely quantum mechanical, and is obtained from the 3-level quantum approximation to EPR arrangements [29, 30, 31], $keto-amino \rightarrow enol-imine$ [35, 36, 37, 38, 39, 49, 50, 54]. Classical considerations of biological information processing treat molecular informational dynamics in terms of arrangements and rearrangements of classical “ball-and-rod” molecular units that store and process classical information digital bits [28, 43, 44, 45, 115]. But quantum informational content [16, 17, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45] embodied within entangled proton qubit superpositions (Figure 2-4, Table 2) – $G'-C'$, $G'-C'$, $A'-A'$ – occupying intramolecular decoherence-free subspaces [11, 67, 68, 69], require enzyme – proton entanglement processing, where a proton qubit eigenstate [35, 36, 37, 38, 39] is quantum mechanically selected [11, 40, 41, 42, 43, 44, 45] to specify observable ts or td [15, 16, 17, 35, 36, 37, 38, 39, 49, 50, 54].
Robust Homo sapiens inherit normal, evolutionarily conserved “cancer genes” [55, 56, 75, 140, 141, 142, 143], “Alzheimer’s genes” [76, 77, 144, 145, 146] and the huntingtingene [37, 53, 54, 109, 110], each of which is associated with its “wild-type” CNGS, $s$ [113, 114], defined, approximately, by the inequality, $1 \geq s \geq 0.97$ [35, 36, 37, 38, 39, 49, 50]. This model considers three different sets of Q individuals ($Q \geq 100,000$) the populations who have inherited a normal target domain, $s$ ($1 \geq s \geq 0.97$), which includes conserved “cancer genes”, “Alzheimer’s genes” and the huntingtin gene. After developing the EPR-entanglement Darwinian polynomial, each of the three age-related genetic diseases is evaluated for genotypic and phenotypic expression, as a function of classical and quantum entanglement genetic contributions, to each of the three respective genes.
A general expression for the total biological noise, $N(t)$, in all $Q$ individual genes, $g$, in the population at age $t$ is given by
$$N(t) = Q\left\{N_0 + \sum_{i=1}^{m} \lambda_i t + \sum_{j=1}^{k} \left(\beta_j / 2\right) t^2\right\} \tag{18}$$
where $N_0$ is the average number of mutations originating by classical and entanglement channels – per gene $g$ in the population of $Q$ at $t = 0$. The sum $\sum_{j=1}^{m}$ is over all $m$ G-C + A-T pairs in the relevant gene where mutations originate by classical Newtonian operations on DNA [28, 71, 139]. The sum $\sum_{j=1}^{k}$ is over the $k$ G-C + A-T pairs in the gene (generally, $m = k$) where quantum uncertainty limits, $\Delta x$ $\Delta p_x \geq \hbar/2$, are imposed on metastable hydrogen bonding amino protons, creating confined spaces, $\Delta x$, which cause direct quantum mechanical proton – proton physical interaction. This generates probabilities of EPR arrangements, $keto-amino \rightarrow enol-imine$, such that position and momentum entanglement is introduced between separating enol and imine protons [29, 30, 31, 35, 36, 37, 38, 39, 54]. Product enol and imine protons are shared between two indistinguishable sets of electron lone-pairs, belonging to enol oxygen and imine nitrogen in decoherence-free subspaces [11, 67, 68, 69] on opposite strands, and consequently, participate in entangled quantum oscillation between near symmetric energy wells (Figure 20, Tables 8-9, Appendix II) at $\sim 4 \times 10^{13} \text{ s}^{-1}$ until “measured by”, $\delta t << 10^{-13} \text{ s}$, QPIE [35, 36, 37, 38, 39, 40]. The EPR-entanglement algorithm yields molecular clock $ts$ and $td$, after ($i$) an initial formation of enzyme-proton entanglement, $\delta t << 10^{-13} \text{ s}$, ($ii$) implementation of an entanglement-assisted enzyme quantum search ($\Delta t' \leq 10^{-14} \text{ s}$), ($iii$) specification of the “correct” complementary mispair (Figure 6), and ($iv$) selected replication-substitution or deletion [15, 16, 17, 35, 36, 37, 38, 39, 54], with classical tautomers containing decohered protons. The $\beta t^2$ term in Eq (18) is obtained from a 3-level quantum approximation to EPR arrangements [49,50; Appendix I], $keto-amino \rightarrow enol-imine$. However, $\sum_j \beta t^2$ terms are experimentally contributing observables if and only if quantum entanglement processes ($i$) through ($iv$) above are properly executed by the enzyme quantum processor [35, 36, 37, 38, 39, 40].
Consistent with observation [35, 36, 37, 38, 39, 55, 75, 76, 77, 140, 141, 142, 143, 144, 145, 146], this model assumes that target genes can because of accumulating an evolutionarily defined level of EPR-generated entangled proton qubits (stochastic mutations [15, 16, 17, 35, 36, 37, 38, 39, 100, 101, 102, 103, 104]) plus classical “point” mutations be “converted” into a disease producing mode. The time rate of change of converted target genes, $dg(t)/dt$, is proportional to the total number of entangled proton qubits in the relevant genetic domain plus classical replication-dependent Newtonian mutations contained in each of the $Q$ genes, $g(t)$, in the population at age $t$. This is given by
$$\frac{d}{dt} g(t) = 1/K N(t) \tag{19}$$
where $1/K$ is the proportionality constant, and $N(t)$ is the noise defined in Eq (18). The number of converted target genes, $g(t)$, in the population of $Q$ at age $t$ is given by
$$g(t) = g_0 + Q/K \left\{N_0 t^2 + \sum_{i=1}^{m} \left(\lambda_j / 2\right) t^2 + \sum_{j=1}^{k} \left(\beta_j / 6\right) t^3 \right\} \tag{20}$$
where $g_0$ is the number of converted genes in the population at $t = 0$. Phenotypic expression incidence, $E(t)$, in the population of age $t$ would change at a rate, $dE/dt$, which is proportional to the total number of converted genes, $g(t)$, in the population. This relationship is expressed as
$$\frac{d}{dt} E(t) = \frac{1}{B} g(t) \tag{21}$$
where $1/B$ is the proportionality constant. The incidence of phenotypic expression, $E(t)$, in the population at age $t$ is given as
$$E(t) = E_0 + \left(\frac{g_0}{B}\right) t + Q/2KB \left\{N_0 t^2 + \sum_{i=1}^{m} \left(\lambda_j / 3\right) t^3 + \sum_{j=1}^{k} \left(\beta_j / 12\right) t^4 \right\} \tag{21}$$
where $E_0$ is the incidence at $t = 0$. Here time $t = 0$ when the egg is fertilized. If the $s$-domain in the “cancer gene” or “Alzheimer’s gene” were populated by entangled proton qubits to its threshold limit at conception, i.e., to $s \approx 0.97 + \varepsilon$, the model implies spontaneous abortion would be a consequence [35, 151]; so, in these cases, a live birth implies $E_0 = g_0 = 0$ at $t = 0$ in Equation (22). Therefore, $N_0$ is the average number of inherited mutations per gene, including classical and entanglement-originated $ts + td$ accumulated in CNGS in prior generations. Entangled proton qubit states per se are not inherited [35, 36, 37, 38, 39, 64], but accumulate with time at rates governed by quantum uncertainty limits, $\Delta x \Delta p_x \geq \hbar/2$, operating on metastable hydrogen bonding amino DNA protons [65].
Manifestation of Huntington’s Disease in terms of EPR-Entanglement and Classical Contributions to Expanded (CAG)n Repeats within the Huntingtin Gene
k In the case of Huntington’s disease [37, 53, 54, 109, 110], an increase in length of an inherited (CAG)n repeat is represented in Equation (22) by an appropriate increase k =∑ .
t β of the upper limit, k, on the sum over j terms, 4
j j
1 These contributions to E(t) are coefficients of t4 terms; so, as the upper limit, k, would increase for longer (CAG)n repeats, contributions to E(t) would increase nonlinearly.
m =∑ , are summed over all m base t λ
3 Classical terms,
i i
1 pairs of the huntingtin gene including the embedded (CAG)n component — which, generally, would be significantly longer than the inserted (CAG)n repeat. In this case, changes in (CAG)n repeat length would m =∑ t λ
3 represent relative small fluctuations in i i
1 contributions to E(t) in Equation (22). Thus, nonlinear contributions to E(t) as inherited (CAG)n repeats become longer [35, 37, 53, 109], illustrated in Figure 14, are consistent with consequences of EPR-generated entangled proton qubits populating expanded (CAG)n repeats which, in Equation (22), contribute as coefficients of t4 terms.
![Figure 18: Evolution of genomic lineages from the primordial gene pool. ((from Figure 2, Koonin et al. [61] with permission). Characteristic images of RNA and protein structures are shown for each postulated stage of evolution, and characteristic virion images are shown for emerging classes of viruses. Thin arrows show the postulated movement of genetic pools between inorganic compartments. Block arrows show the origin of different classes of viruses at different stages of pre-cellular evolution.) A reverse-time extrapolation from observables [15-21] exhibited by ancient T4 phage DNA [46] implies that entangled proton qubits could have originally emerged in the first “susceptible” ancestral duplex segments of primitive RNA – ribozyme systems [58-61]. This assumption requires primordial ribozyme – RNA duplex segments to simulate, approximately, conditions exhibited by ancient T4 phage DNA systems that accumulate EPR-generated entangled proton qubits in metabolically inert (extracellular [64], pH 7, 20 0C) base pair isomer superpositions, observed as _G′-C′, *G-*C_ and _*A-*T_ [15-23]. This scenario provides a possible source of “RNA-type” hydrogen bonded duplex molecules [28,65] susceptible to occupancy by EPR-generated [29-31] entangled proton qubits [15-17,35-39,54,104], and thus, allowed ancestral peptide-ribozyme – RNA systems [57- 61] to form entanglement states with oscillating, │+>⇄ │–>, entangled proton qubits, occupying decoherence- free subspaces [11,67-69] within hydrogen bonded [65] duplex RNA segments [35,36]. At this stage of evolutionary development, peptide-ribozyme – proton entanglements could implement an entanglement- directed quantum search, Δt′ ≤ 10‒14 s < τD< 10−13 s [13], to select the next amino acid electron lone-pair, or amino proton, to be added to the pre-protein peptide polymer [35,36]. Additionally before proton decoherence, τD< 10−13 s, operations of the entangled ribozyme – proton system included (_a_) generating a transcribed message based on quantum informational content of “measured” entangled proton qubits [15-17,37-39], (_b_) implementing an entanglement-directed quantum search, Δt′ ≤ 10_−14_ s [13], that specifies the incoming base’s electron lone-pair, or amino proton, for evolutionary substitution, **_ts_**, or deletion, **_td_**, and (_c_) feedback responding “Yes” or “No” to _translation_ of the transcribed “qubit message” [16,17]. “Yes” implies existence of an “_r__+__-_type” allele which allows replication initiation, but “No” identifies an unacceptable “mutant allele”, and therefore, replication is denied. Before “DNA-type” repair enzymes were introduced [139,154-155], RNA – ribozyme systems avoided evolutionary extinction by disallowing conserved, “_r__+_- type” allele contribution to the gene pool when “excessive” EPR-generated mutations (entangled qubits) were present [23,35,36]. Other “relaxed” genes were allowed mutation, variation and selection. These processes introduced viable peptide-ribozyme – RNA, wild-type “_r__+_-systems” where peptide-ribozyme – proton entanglements were exploited to generate rudimentary peptide chains that subsequently usurped ribozyme functions. When duplex RNA genomes became “too](/fulltextimages/1462/fig_18.jpeg)
Since unperturbed β is small (~ 2×10−23 sec−2; Cooper, unpublished results), Equation (22) demonstrates that =∑ contributions to E(t) could be effectively zero for t β
4 j j
1 long time periods if the upper limit on k is small, i.e., short (CAG)n repeats. For example, humans that inherit (CAG)n repeats with n ≤ 36, do not exhibit Huntington’s disease in their generation [109], but subsequent generations who inherit (CAG)n with n ≥ 37 can manifest disease condition in later life [35, 37, 53, 109]. Also, agreement between observation (Figure 14) and the model [37, 54] implies k =∑ t β that “nonlinear” data are consequences of 4 j j
1 m =∑ , t λ
3 contributions. If classical components,
i i
1 contribute to E(t) in Equation (22), genotypic inheritance of long (CAG)n (n ≥ 70) repeats could be immediately (hrs., days, weeks) expressed at birth by classical [28, 57, 71, 139] transcription and replication, which is not the case. However, all inherited (CAG)n base pairs would, at t = 0, initially contain recently replicated metastable keto-amino hydrogen bonds [35, 36, 37, 38, 39, 64], but infants who inherit very long (CAG)n repeats, e.g., n ≥ 70, do not exhibit disease at birth. The inherited, expanded (CAG)n, e.g., n ≥ 70, sequence of metastable keto-amino hydrogen bonds provides a sizable cross-section, susceptible to EPR-generated entangled proton qubits [35, 36, 37, 38, 39, 54]. In the case of the huntingtin gene, if the s-domain (1 ≥ s ≥ 0.97 + ε) within a large (CAG)n repeat, e.g., for n = 70, of the huntingtin gene were populated by entangled proton qubits to its threshold limit at conception [49], i.e., to s ≈ 0.97 + ε, the model implies spontaneous abortion would result [35, 37, 151]. Figure 14 data imply a live birth who had inherited a (CAG)70-repeat within the huntingtin gene would exhibit “normality” for ~ 2 to ~ 12 years, before displaying phenotype [53]. The time between birth and manifestation of Huntington’s disease (Figure 14) is the time required for EPR-generated entangled proton qubits to populate the (CAG)n sequence to its minimal “threshold limit” [49, 50, 64], and subsequently, exhibit phenotypic expression [35, 37, 54] as consequences of quantum processors [40] “reading” quantum informational content embodied within EPR-generated entangled proton qubit states [35, 36, 37, 38, 39]. Note in Figure 14, age-of-onset for n = 70 and n ≈ 86 is approximately identical, i.e., age ≈ 2 y. This is consistent with the concept that inherited (CAG)n-repeats (where n = 70 or n = 86) must become populated to its respective “threshold limit” [37] with EPR-generated entangled proton qubits, which are subsequently measured by Grover’s [40] “transcriptase” quantum processor, and consequently, phenotypic expression is exhibited [53, 54]. This observation implies that $\sim 2$ y, after birth, are required to populate the “threshold limit” of long (CAG)$_n$-repeats, i.e., $n \geq 70$, with EPR-generated entangled proton qubits.
In the case of very long inherited (CAG)$_n$ repeats ($n \geq 70$), the phenomenon of genetic anticipation is exhibited where earlier onset, and more severe disease, is manifested due to entangled proton qubits populating the expanded (CAG)$_n$ sequence beyond its “minimal” threshold limit for smaller (CAG)$_n$ sequences [37]. This explanation is also applicable to children who inherit congenital myotonic dystrophy (CDM) in the form of long (CTG)$_n$ ($n \geq 750$) repeats [111, 152]. In these cases, phenotypic expression of CDM is not exhibited until $\sim 1$ year after birth. Based on agreement between observation (Figure 14) and quantum theoretical predictions [1, 2, 3, 37]
$$\sum_{j=1}^{k} \beta_j t^4$$, in Equation (22) phenotypic expression of Huntington’s disease requires enzyme quantum processor measurements [40] of EPR-generated entangled proton qubit states; otherwise, Huntington’s disease would be “immediately” (hours/days/weeks) expressed after birth by classical Watson-Crick-Muller transcription and/or replication of expanded (CAG)$_n$ ($n \geq 70$) repeats containing metastable keto-amino hydrogen bonds [28, 65, 71], which is not observed [53].
Clearly, the enzyme quantum reader distinguishes genetic information embodied within a classically originated base pair, e.g., G-C [28], from an entanglement-originated $G'-C'$ superposition (Table 2) [35, 36, 37, 38, 39], consisting of 16 different entangled proton qubit states, Equation (6). Evidently, manifestation of Huntington’s disease [53], and analogously, manifestation of CDM [111, 152], requires quantum processor [35, 36, 37, 38, 39, 40] measurements of EPR-generated [29, 30, 31, 32, 33, 34] entangled proton qubit states, occupying a “threshold limit” [37, 109], to exhibit phenotypic expression of these genotypically inherited (CAG)$_{70}$ and (CTG)$_n$ ($n \geq 750$) repeat diseases [37, 53, 111, 152]. Consistent with observation [53], the contribution to phenotypic expression of Huntington’s disease is simulated by nonlinear, quantum entanglement terms, $$\sum_{j=1}^{k} \beta_j t^4$$, in Equation (22). Also, if life expectancy were to significantly increase, disease-free copy numbers [109] of (CAG)$_n$ and (CTG)$_n$ [111, 152] would necessarily become smaller [37].
Age-Related Tumorigenesis in Terms of the EPR-Entanglement Darwinian Polynomial
Annual incidence data (Figure 15) on the 74 “class 1” tumors identified by the Connecticut Tumor Registry between 1968 and 1972 generate the empirically determined equations [56].
Log (percentage total incidence) = 0.031 (age) – 1.15 (23) and Log (percentage total incidence) = 0.027 (age) – 0.897 (24)
for males and females, respectively (ages 10 to 80 y). The exponential behavior of Eqs (23-24) is displayed in Figure 15, where average percentage total incidence as a function of age is proportional to $t^4$, and differences between male and female incidence curves in Figure 15 are negligible. “Class 1” tumors are identified as those that exhibit a single incidence peak at age > 50 y, whereas “class 2” tumors (e.g., bone, lymphatic leukemia, testis and Hodgkin’s disease; data not shown) exhibit two incidence peaks; one at age < 35 y and one at age > 50 [56]. The ~70% increase in stomach cancer observed in white males, ages 25 to 39 y, over three decades, 1977 to 2006 [142] is an enigmatic “class 2” manifestation of cancer. Childhood cancer [56] is expressed at age $\leq 10$ y and, thus, is a special case of “class 2” tumors.
The EPR-entanglement Darwinian polynomial, Equation (22), describes “point” mutational events originating via classical [28, 71, 139] and quantum entanglement algorithmic processes [35, 36, 37, 38, 39, 40, 54, 100]. Classical Newtonian operations are simulated by $$\sum \lambda t^3$$ terms, whereas $ts$ and $td$ are consequences of the quantum entanglement algorithm, i.e., entangled proton qubits accumulated in decoherence-free subspaces [11, 67, 68, 69] that are processed by enzyme – proton entanglement measurements [15, 16, 17, 35, 36, 37, 38, 39, 40]. The resulting $ts$ and $td$ are “end-product” contributions expressed by quantum entanglement terms, $$\sum \beta_j t^4$$, in the entanglement Darwinian polynomial, Equation (22). If, however, EPR-generated, entangled proton qubits in decoherence-free subspaces were not physically available, time-dependent quantum informational content would not exist for enzyme – proton entanglements to process [40, 41, 42, 43, 44, 45], yielding observable quantum entanglement terms, $$\sum \beta_j t^4$$ in Equation (22) [35, 36, 37, 38, 39].
in Equation (22), would not simulate empirically generated cancer incidence data, which per Figure 15, is contrary to fact. Thus, quantum
4 j j t β ∑ , efficiently entanglement theoretical terms, simulate cancer incidence data as a function of age (Figure 15), implying EPR arrangements, keto-amino → enol-imine, introduce entangled proton qubits into human genomes, which are efficiently deciphered by enzyme – proton entanglements [40, 41, 42, 43, 44, 45] to yield ts and td, exhibited by T4 phage [15, 16, 17], human gene systems [35, 37, 39, 49, 50, 51, 54, 104] and evolving human-rodent microsatellites
4 j j t β ∑ terms and [35, 38, 52]. Agreement between Figure 15 data implies that age-related malignant genotype is efficiently simulated by the intrinsic, evolutionarily selected quantum entanglement algorithm responsible for expressing time-dependent mutations, ts and td [15, 16, 17, 35, 36, 37, 38, 39, 49, 50, 54], whereas classically $$ \sum_ {j} \beta_ {j} t ^ {4} \mathrm {d o n o t} $$ originated mutations [28, 57, 115, 139]
satisfy the ~ t4 age-related manifestation of malignancy exhibited in Figure 15.
Analysis implies Homo sapiens genomic systems distinguish entanglement-originated ts [35, 36, 37, 38, 39] G′2 0 2 → T, G′0 0 2 → C, *G0 2 00 → A, *C2 0 22 → T (“driver” mutations [55, 75, 140]) from classical “repair” substitutions [139] G → T, G → C, G → A, C → T, etc. (“passenger” mutations) introduced by Newtonian operations on DNA [28, 71]. This and other reports [35, 36, 37, 38, 39, 49, 50] imply that evolutionary differences between “driver” and “passenger” mutations [55, 75, 140] are consequences of their very different quantum entanglement and classical evolutionary origins (see Tables 1&2), which are communicated at time of enzyme quantum reader “measurements” [35, 36, 37, 38, 39]. In these molecular genetic genomic operations [56], genetic specificity of a Homo sapiens base pair is governed by (a) chemical composition and (b) its quantum entanglement [7, 8, 9, 10, 11, 35, 36, 37, 38, 39] or classical [28, 71, 139] properties at time of enzyme quantum reader measurement, δt << 10–13 s. Compatibility between observation, Figure 15 [56], quantum theory [1, 2, 3, 4, 29, 30, 31, 32, 33, 34], and evolution of “conserved genes” [35, 36, 37, 38, 39, 53, 54, 55, 56, 75, 76, 77, 78, 79, 104, 105, 106, 107, 108, 109, 110, 111, 140, 141, 142, 143, 144, 145, 146] clearly relates age-related disease manifestation to natural, evolutionarily acquired abilities of “quantum readers” [40] to implement enzyme – proton entanglement processing of EPR-generated entangled proton qubits, thereby probabilistically creating consequences of evolutionarily selected ts and td. In the case of age-related cancer [56], evolutionarily acquired bio-physical properties of cells are responsible for implementing the EPR-entanglement algorithm that generates age-related genotypic expression of disease $$ \sum_ {j} \beta_ {j} t ^ {4} $$
, but classical “ball-and-rod” Newtonian [35, 36, 37, 38, 39], operations [28, 57, 71, 139] do not contribute to the resulting “driver” mutation spectra of Figure 15. Agreement between Equation (22) and Figure 15 implies the quantum entanglement algorithm introduces cancer causing “driver” mutations [35, 39, 55, 75, 140, 141], ts expressed as SNPs where the enzyme quantum reader distinguishes quantum informational content, yielded by dynamic entangled proton qubits [35, 36, 37, 38, 39], from classically introduced [28, 57, 71, 136], “passenger” mutations [55, 140]. In this case, age-related cancer [56] exhibited in Figure 15, is a consequence of normal, evolutionarily selected quantum entanglement algorithm processes, $$ \sum_ {j} \beta_ {j} t ^ {4} $$
, designed to preserve a “wild-type” form of gene pool viability [35, 36, 37, 38, 39].
Origin of Late-onset Alzheimer’s disease in terms of the EPR-Entanglement Darwinian Polynomial
When CNGS, s (1 ≥ s ≥ 0.97) [49, 50, 113, 114], accumulate entangled proton qubits to an “unsafe” threshold, i.e., to s ≈ 0.97 + ε, “gatekeeper” genes [35, 36, 37, 38, 39] implement their evolutionary function of discriminating against “unsafe” genomes. Consequently, “wild type” gene pool viability is evolutionarily preserved, which enables species’ survival at the expense of sacrificing operational, but “unsafe” entangled “proton qubit-depleted” host, haploid and diploid genomes [37, 38, 39, 56, 77, 151]. After CNGS have “benignly” accumulated entangled proton qubit mutations ts + td to the “threshold limit”, s ≈ 0.97 + ε [49], the probability is enhanced that subsequent ts introduce SNP mutant proteins responsible for eliminating “unsafe” genomes. “Cancer genes” [55, 75, 140, 141, 142, 143] and “Alzheimer’s genes” [76, 77, 144, 145, 146] are two example “gatekeeper” gene systems that exhibit similar [56, 77] age-related manifestation of a lethal disease as consequences of entangled proton qubit accumulation to an “unsafe” threshold, i.e., to s ≈ 0.97 + ε. In these cases, entangled proton qubit accumulations are deciphered by QPIE which introduce SNPs ts that specifically express selected mutant proteins to manifest an age-related lethal disease [35, 37, 38, 39, 55, 76, 77]. In cases of “cancer genes” [55, 75, 140, 141, 142, 143] or identifiable “Alzheimer’s genes” [76, 77, 144, 145, 146] e.g., APOE, APP, PSEN1 and PSEN2 ts are expressed as SNPs which can cause disease, or a significant enhancement of “risk factors” for disease. Also “unperturbed” age-related manifestation of each disease exhibits exponential increases in disease incidence [56, 77] as a function of age (Figure 15-16). Due to acquiring a “threshold limit” of entangled proton qubits [35, 36, 37, 38, 39, 49, 50], conserved “gatekeeper” genes, e.g., “cancer genes” and “Alzheimer’s genes”, exhibit their evolutionarily selected, age-related lethal maladies, which discriminate against genomes that have acquired entangled proton qubits to “unsafe” levels. Conserved “gatekeeper” genes exhibit their selected, time-dependent lethal functions after entangled proton qubits have accumulated to an “unsafe” threshold [35, 36, 37, 38, 39, 49, 50, 56, 76]. This condition allows the “next” set of ts to introduce SNP mutant proteins that manifest an evolutionarily selected degenerative disease [55, 56, 75, 76, 77, 140, 141, 142, 143, 144, 145, 146]. Unsafe haploid genomes are eliminated during oogenesis or spermatogenesis, thereby preserving CNGS across mouse-rat-human evolution [113, 114], which also prevents “rapid” evolutionary extinction [35, 36, 37, 38, 39, 151]. These similarities in genome “preservation operations” [153] imply that age-related Alzheimer’s disease satisfies an evolutionarily selected function analogous to age-related cancer [56, 77]. Both age-related maladies have been evolutionarily selected to remove, from the gene pool, haploid and diploid genomes that contain “unsafe” levels of entangled proton qubits [35, 36, 37, 38, 39] occupying different CNGS [75, 76, 77, 140, 141, 142, 143, 144, 145, 146]. When an inherited CNGS is significantly populated, e.g., to s ≈ 0.98 + ε at conception, both cancer [56, 142] and Alzheimer’s disease [77, 144, 145, 146] can consequently exhibit early- onset Mendelian inheritance, but unlike cancer, childhood Alzheimer’s disease is not exhibited, i.e., disease expression at age ≤ 10 y.
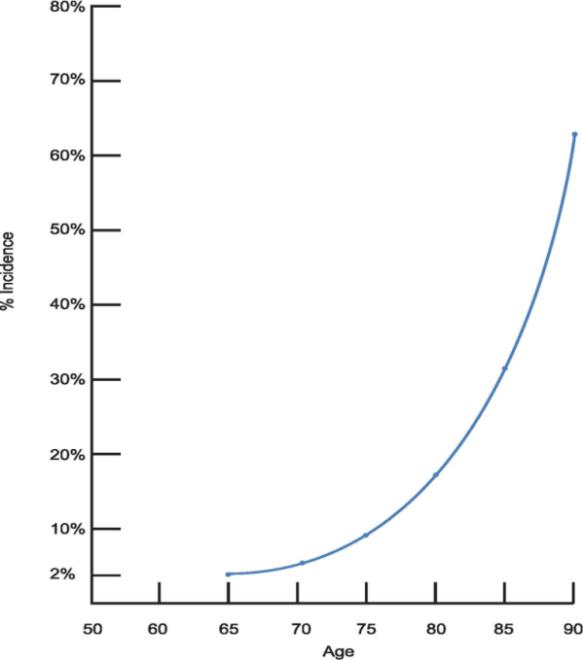
Early-onset Tumors and Alzheimer’s Disease via the EPR-Entanglement Darwinian Polynomial
Based on analyses [35, 36, 37, 38, 39, 49, 50, 54] and observation [56, 76, 77, 142], when the “gatekeeper” condition, s ≈ 0.97 + ε, is satisfied, an evolutionarily determined CNGS, s (1 ≥ s ≥ 0.97), has been populated by entangled proton qubits to its “unsafe” threshold. Subsequent enzyme – proton entanglement “measurements” of quantum informational content embodied within entangled proton qubits yield ts and td; so, consistent with species preservation, genomes with “unsafe” levels of entangled proton qubits are evolutionarily eliminated by mutant proteins generated by evolved “cancer genes” [55, 75, 140, 141, 142, 143] or evolved “Alzheimer’s genes” [76, 77, 144, 145, 146]. Both cancer and Alzheimer’s disease exhibit a small percentage of early-onset disease manifestation, i.e., age < 40 for cancer [56, 142] and age < 55 for Alzheimer’s disease [76, 145, 146]. Also, several childhood cancers exhibit high incidence peaks at age ≤ 10 y [56], but childhood Alzheimer’s disease is not observed.
In terms of Equation (22), inherited ts and td that occupy CNGS, s (1 ≥ s ≥ 0.97), and thus could be expressed as childhood Alzheimer’s disease would cause elimination of those genomes during spermatogenesis and oogenesis, but inherited ts and td that contribute to childhood cancer do not cause analogous genome elimination. Early-onset tumors studied by Dix et al. [56] e.g., bone, lymphatic leukemia, testis and Hodgkin’s disease exhibited an initial “high” incidence peak at age < 35 and a second peak at age > 50 y. A more recent study [142] identifies an initial “high” incidence peak of stomach cancer for ages 25 to 39 y. These early-onset cancers particularly childhood cancer can be a consequence of an inherited, considerably populated CNGS, e.g., s ≈ 0.97 + 10ε, for childhood cancer.
Additionally, perturbations that increase energy density of duplex DNA would introduce more energetic vibrational modes (elevated E3 values in Figure 19, Appendix I) [132], thereby elevating energy shift, γp, values, i.e., γp = $\left[ \left( E_3 - E_p \right)^2 / 4 + \alpha_p^2 \right]^{1/2}$ (see Appendix I), which would introduce larger β values into Equation (22). This would enhance EPR arrangement rates, keto-amino → enol-imine, and reduce times required for ts and td to populate CNGS to a threshold limit. The observed ~ 70% increase in stomach cancer among white males, ages 25 to 39 y, over three decades, 1977 to 2006 [142], implies relevant diploid thresholds were populated to their limits by entangled proton qubits at “early” ages. Based on the model, early-onset incidence peaks at age < 39 are consequences of (a) “preventable” perturbations causing increases in energy density of “local” DNA in the existing diploid genome, or (b) this population inheriting a particular set of CNGS that were > 50% populated in the haploid genome of the previous generation, e.g., s ≈ 0.98 + ε at conception.
Agreement between Figure 15 data and $\sum_j \beta_j t^4$ terms in Equation (22) implies “normal” unperturbed CNGS are populated by entangled proton qubits to a threshold limit at an “averaged rate”, consistent with a “smooth” ~ t4 curve for incidence of cancer as a function of age, exhibiting a single “high” incidence peak at age > 50. If, however, “local” energy density of DNA were enhanced by “perturbations”, vibrational modes become more energetic [132, 133] and values for β become larger. This would increase rates of entangled proton qubits populating CNGS to a threshold limit, which could cause “high” initial incidence peaks for age < 39. Data showing ~ 70% increases in stomach cancer over 3 decades for white males, ages 25 to 39 [142], are consistent with a “premature” populating of CNGS [55, 56, 140, 141, 142, 143].
Thus the “smooth” ~ t4 incidence data (ages 10 to 80 y) imply “normal”, entangled proton qubit contributions, $\sum_j \beta_j t^4$, whereas data exhibiting an “early” initial high incidence peak [142] imply (a) “preventable” perturbations in the existing diploid generation, or (b) an inherited set of CNGS that were > 50% populated in the previous haploid generation. In either case, the CNGS s (1 ≥ s ≥ 0.97) model [35, 36, 37, 38, 39, 49, 50] allows expression of conserved “gatekeeper genes” as consequences of entangled proton qubits populating space, s, to its threshold limit, i.e., to s ≈ 0.97 + ε.
This model provides an internally consistent, quantum entanglement algorithm explanation for “early” and “late” manifestation of cancer [56, 142] and, by analogous evolution arguments [35, 36, 37, 38, 39], Alzheimer’s disease [76, 77, 144, 145, 146]. Results of Cruchaga et al. [76] demonstrate that the same genes sensitive to SNPs contribute to both early- and late-onset manifestations of Alzheimer’s disease, which is analogous to the situation exhibited by tumors [55, 140, 141, 142, 143]. Reduced “gatekeeper” genetic spaces, s, could be inherited by progeny of those exhibiting cancer at “early” ages, e.g., age < 39, [56, 142] or Alzheimer’s disease at age < 55 [76, 145, 146].
Orgel’s “Error Catastrophe” Hypothesis via EPR-Generated Entangled Proton Qubits and “Gatekeeper” Genes
Over the past ~ 3.5 or so billion years, pre-cellular [35, 36, 58, 59, 60, 61], prokaryotic [15, 16, 17, 18, 19, 20, 21, 22, 23, 27, 28, 82, 83, 156, 157, 158] and eukaryotic [28, 37, 38, 39, 57, 100, 101, 102, 103, 112, 138] evolution had ample opportunity to select preferable, advantageous mechanisms for protecting the gene pool and conserved noncoding genomic spaces (CNGS) [113, 114] against acquiring unsafe levels of entangled proton qubits in haploid and diploid genomes. A sensitive CNGS [113, 114], s, for Homo sapiens can be defined by the inequality, 1 ≥ s ≥ 0.97 [35, 36, 49, 50]. This is based on 100 y as the maximally allowed Homo sapiens age, and experimental measurements of mean lifetimes, τ, of metastable keto-amino G-C states in genomic DNA (37 °C; pH 7), τ ≥ 3000 y.
(Tables 5-6, Appendix II) [17]. Thus, at age 100 y, ~ 3% (100/3000×100) of G-C sites in the Homo sapiens genome would have been populated by entangled proton qubits, generated by EPR arrangements, keto-amino → enol- imine, via the symmetric and asymmetric channels, G-C → G′-C′ and G-C → *G-*C (Figure 2-3). Subsequent enzyme – proton entanglement processing introduces ts (and td at *A-*T pairs). Accordingly, robust Homo sapiens infants inherit a normal “wild type” CNGS inequality, 1 ≥ s ≥ 0.97, which becomes “uneventfully” populated by entangled proton qubits to its threshold limit, s ≈ 0.97 + ε, at an advanced age. Existence of CNGS across the era of mouse-rat-human [129] implies the condition, s ≈ 0.97 + ε [35, 38, 49, 50], in haploid noncoding DNA segments would result in their elimination during oogenesis and spermatogenesis [130]. Evolutionary elimination of genomes exhibiting consequences of unsafe entangled proton qubits provides an entanglement-enabled mechanism for preserving CNGS across the mouse-rat-human evolution spectrum, ~ 70×106 y [129]. Ultimately, QPIE “measure” [15, 16, 17, 35, 36, 37, 38, 39, 100, 101, 102, 103, 104] quantum informational content of entangled proton qubits to yield molecular clock ts and td. Manifestation of ts and td requires (i) an initial formation of enzyme-proton entanglement (δt << 10−13 s), (ii) implementation of an entanglement-assisted enzyme quantum search (Δt′ ≤ 10_−14_ s), (iii) specification of a “correct” complementary mispair, and (iv) selected replication-substitution or deletion [15, 16, 17, 35, 36, 37, 38, 39], with classical tautomers containing decohered protons. The resulting ts are exhibited as G′2 0 2 → T, G′0 0 2 → C, *G0 2 00 → A_and *C2 0 2_2 → T which are generally expressed as SNPs [15, 16, 17, 35, 36, 37, 38, 39, 100, 101, 102, 103, 104], whereas td are consequences of *A-*T site deletions (Figure 4) [16, 38]. Observation [15, 16, 17, 18, 19, 20, 21, 55, 56] and theory [35, 36, 37, 38, 39, 54] imply quantum entanglement algorithm mechanisms responsible for ts, cancer causing “driver” mutations [35, 36, 49, 50, 55, 75, 140, 141, 142, 143], are biologically distinguishable from classical [27, 28, 57, 71, 139] Newtonian operations on DNA that yield benign, “passenger” base substitutions [55, 75]. Although DNA repair enzymes [139] were acquired during the transition from ancestral RNA to DNA genomes [35, 36, 155], the originally selected quantum entanglement algorithm for RNA genomic evolution was retained, and further refined, for entanglement-originated ts and td in DNA systems. Consequently, all stages of genomic DNA evolution precellular [35, 36, 58, 59, 60, 61], prokaryotic, [15, 16, 17, 18, 19, 20, 21, 27, 28, 36, 139, 156, 157, 158], eukaryotic [37, 38, 39, 57, 138] were successfully executed under conditions of continuous accumulations of entangled proton qubits, subsequently deciphered by enzyme – proton entanglements to yield ts and td, exhibited as SNPs [15, 16, 17, 18, 19, 20, 21, 22, 23, 35, 36, 37, 38, 39, 54, 100, 101, 102, 103, 104]. The continuous acquisition of entangled proton qubits provides an evolutionary rationale for “protection” against consequences of “unchecked” accumulations of entangled qubits that would be “unsafe” if contributed to the gene pool [35, 36, 37, 38, 39, 49, 50, 151]. To this end, conserved “cancer genes” [55, 75, 140, 141, 142, 143], “Alzheimer’s genes” [76, 77, 144, 145, 146], the huntingtin gene[37, 53, 54], and “other” genes [78, 79, 159] emerged evolutionarily under conditions of continuously accumulating entangled proton qubits that were enzymatically deciphered to yield entanglement- generated ts and td, exhibited as SNPs. Consistent with the present and other reports [35, 36, 37, 38, 39], evolutionarily conserved “cancer genes” [55, 75, 140, 141, 142, 143] and “Alzheimer’s genes” [76, 77, 144, 145, 146] express their respective age-related maladies as consequences of quantum entanglement algorithmic processes operating on normal, EPR-generated entangled proton qubits [15, 16, 17, 29, 35], acquired beyond “unsafe” CNGS (1 ≥ s ≥ 0.97) threshold limits of s ≈ 0.97 + ε[36, 37, 38, 39, 49, 50]. When the “threshold” condition s ≈ 0.97 + ε is satisfied, the probability is significantly enhanced that the subsequent round of ts and td will express selected SNP mutant proteins [35, 36, 37, 38, 39, 55, 75, 76, 77, 140, 141, 142, 143, 144, 145, 146] responsible for disease manifestation. If conserved “gatekeeper” genes had been populated to the threshold limit, i.e., to s ≈ 0.97 + ε, and were then contributed to the gene pool, rapid evolutionary extinction would ensue [16, 54, 151]. Consequently, genomes containing CNGS populated to “unsafe” threshold limits, by entangled proton qubits, are evolutionarily eliminated by normal Darwinian cellular processes [28, 35, 36, 37, 38, 39, 130]. Haploid genomes populated by entangled proton qubits to “unsafe” levels are eliminated during spermatogenesis and oogenesis, thereby preserving “wild type” gene pool viabilities, and enabling species survival [26, 35, 36, 37, 38, 39]. However diploid genomes populated by entangled proton qubits to “unsafe” threshold limits [35, 36, 37, 38, 39, 49, 50] encounter discrimination by conserved “gatekeeper” genes [35], e.g., “cancer genes” [55, 75, 140, 141, 142, 143], “Alzheimer’s genes” [76, 77, 144, 145, 146] and “other” genes [53, 78, 79, 111, 152, 153, 159]. In these cases, ts introduce specific SNPs which manifest the respective age-related degenerative disease [35, 36, 37, 38, 39, 55, 75, 76, 77, 140, 141, 142, 143, 144, 145, 146, 159]. Also, natural selection [28, 35, 36, 57, 100, 101, 102, 103, 138] would eliminate deleterious genes that accumulate “unsafe” entangled proton qubits, including “cancer genes” [55, 75, 140, 141, 142, 143], “Alzheimer’s genes” [76, 77, 144, 145, 146], and the huntingtin gene [35, 37, 53, 54, 109], which did not happen. Rather these evolutionarily conserved “beneficial genes” have been retained to execute their necessary “gatekeeper” functions [35, 37, 38, 39] of disallowing contributions to the gene pool by genomes that have acquired entangled proton qubits to “unsafe” levels. This prevents “unsafe” contamination of the gene pool, which enables species’ survival at the expense of sacrificing an “operational”, but depleted, host genome [16, 35, 36, 37, 38, 39, 49, 50]. Operational functions of “gatekeeper” genes described here provide a quantum entanglement algorithm interpretation of Orgel’s [160] classical “error catastrophe” hypothesis, where quantum uncertainty limits, Δx Δpx ≥ ћ/2, operate on hydrogen bonding amino protons to introduce probabilities of EPR-generated entangled proton qubits. When CNGS, s (1 ≥ s ≥ 0.97), of a “gatekeeper” gene acquire entangled proton qubits to a threshold limit, i.e., to s ≈ 0.97 + ε, such genes are disallowed contributions to the gene pool, due to selected “error catastrophe” criteria [35, 36, 37, 38, 39, 160, 161, 162]. This interpretation of the “error catastrophe” hypothesis, in terms of “gatekeeper” genes [35, 37, 38, 39], and Grover’s [40] processors “reading” quantum information [41, 42, 43, 44, 45] embedded within EPR-generated entangled proton qubits, appears applicable to oocytes that exhibit normal menopause [35, 39, 130]. In this case, normal human menopause [130] is implemented by “gatekeeper” genes [37, 38, 39] that disallow haploid genomes containing “unsafe” levels of EPR-generated entangled proton qubits from contributions to the gene pool. This allows the “gene pool” to retain an approximately “wild-type” spectrum of genes that exhibit age-related disease [35, 37, 38, 39, 53, 55, 75, 76, 77, 140, 141, 142, 143, 144, 145, 146].
Hypothesis: Origin of Molecular “Life- forms” on Prebiotic “Earth-Like” Planets
Credible scenarios [57, 58, 59, 60, 61] for origin of first molecular life on “Earth-like” planets require mechanistic explanations [35, 36] for an “early” prebiotic Earth to acquire and organize complex organic molecules such that molecular evolutionary processes could be successfully implemented. Section II noted that the origin of life hypothesis discussed here is within the context of a “Big Bang” [85, 86] or “Big Bounce” [87, 88] origin (~13.8 by a) of mass, particles, energy, and information embedded within massive particles and energy fields (nuclear, gravitational, thermal, and electromagnetic).
This origination specifies how particles and energy fields self-interact and interact with each other. Incremental evolutionary processes are presented in terms of low energy, quantum entanglement reactivity [7, 8, 9, 10, 11, 29, 30, 31] interfacing with prebiotic protons [35, 36], electron lone-pairs and classical oligomers [37, 38, 39, 58, 59, 60], which involves operational interfaces between entanglement information processing, Δt´ ≤ 10−14 s [13, 35, 36, 37, 38, 39], and decohered classical observables [14] that expedite, and generate, reactive in vivo bio-molecular evolutionary phenomena [35, 36, 37, 38, 39, 54, 100, 104]. Models for origin and evolution of first primitive molecular “life- forms” on planet Earth [58, 59, 60, 61] can be constructed in terms of assumed chemical and physical reactive events [35, 36, 57, 58, 59, 60, 61, 62, 63, 84] that progressively “evolved” through three developmental primordial phases to establish an evolving DNA – protein system [35, 36], which represents an initial origin of sustainable life (OoSL) bio-molecular complex [37, 38, 39, 57]. Initially (Figure 7), ~ 4.3 to 3.9 billion y ago, asteroids and icy comets [89] containing primordial nucleobases [92, 93, 94], long chain polycyclic aromatic hydrocarbons [95], Fullerenes, etc. [96] collided with a cooling prebiotic Earth to create environments conducive to formation of complex organic molecules. Since origin of self-replicating “genome-like” polymers requires existence of informational molecules necessary to initiate self- replication, one can postulate that synthetic processes described by Goldman and Taublyn [89] could have participated in generating precursors for amino acids, polypeptides, small-chain aromatic hydrocarbons, short “RNA-like” polymers, and DNA [58, 59, 60, 61]. Combinations of reactive products within impact environments could incrementally become selectively advantageous for the creation of molecular complexes to synergistically add or incorporate analogous molecular units, and implement primitive polymerization of nucleotides, oligomers and peptides [59, 60, 61]. In these cases, advantageous reactive processes were preferentially selected by environmental conditions. The ensuing “local” environmental impact conditions may have ultimately introduced precursor “RNA-like” polymers, from which primitive, but functional, “ribozyme-like” [58, 59, 60, 61] structures emerged in the primordial pool. Over a period of ~ 300 × 106 y, impact synthetic processes evidently generated “ribozyme-like” RNA polymers that could catalytically link a few molecular units of RNA. Subsequent “ribozyme-like” variants emerged that could more efficiently duplicate RNA “molecular units”. Primordial molecular polymer complexes on prebiotic Earth [59, 60, 61, 82, 83, 84] could generate probabilistic variant systems that occasionally would exhibit improved efficiencies at surviving in their environments. These initial incremental chemical – physical improvements allowed “original” molecular complexes to acquire “RNA-like” polymer structures, e.g., ribozymes [57, 58, 59, 60, 61], which can inefficiently duplicate ~ 10 to 80 or more molecular RNA units per 24 hrs. This nebulous explanation provides a scenario for possible origin of ancient ribozymes [35, 36, 58, 59, 60, 61]. The second developmental phase involves single strand “RNA-like” polymers forming energetically preferable hydrogen bonding [28, 65] duplex loops. Random classical processes [12] allowed keto – amino hydrogen bond formation [35] between energetically preferable, duplex segments of complementary RNA base pairs [28, 36, 59, 60, 61]. When hydrogen bonded amino (‒NH2) protons encountered quantum uncertainty limits [2, 66], Δx Δpx ≥ ħ/2, a probability of EPR-arrangement [29, 30, 31], keto – amino ‒ (entanglement) → enol – imine, was introduced, illustrated in Figure 1-4 for duplex DNA systems, which here is reverse-time extrapolated for applicability in ancestral duplex RNA segments of primordial RNA – ribozyme systems [35, 36, 57, 58, 59, 60, 61]. The resulting EPR-generated [29, 30, 31] reactive processes yielded reduced energy, entangled enol and imine product proton qubits [36, 43] that were each shared between two indistinguishable sets of electron lone-pairs belonging to enol oxygen and imine nitrogen on opposite RNA genome strands [35, 36, 37, 38, 39]. These proton qubits in duplex RNA segments consequently exhibit entangled quantum oscillations, ~ 4×1013 s-1, between near symmetric energy wells (Tables 8 & 10) in intramolecular decoherence-free subspaces [11, 17, 67, 68, 69]. Survival of ribozyme RNA duplex segments containing quantum enhanced information required a “deciphering instrument” [40, 41, 42, 43, 44, 45, 100] that could process and/or repair quantum informational content embodied within entangled proton qubits occupying duplex RNA segments [35, 36]. This was incrementally achieved by exploiting rudimentary peptide-ribozyme entanglement states, with proton qubits, to generate primordial peptide bonds [28, 35, 36, 57, 58]. Ribozyme – proton entanglement states “selected” electron lone-pairs, or amino protons, of primordial amino acids to create “new” peptide bonds between accessible amino acids [57, 58, 59, 82, 83, 84]. Due to peptide chain growth, more efficient rudimentary protein enzymes were introduced, which ultimately usurped ribozyme functions [35, 36]. The resulting RNA – protein systems evolutionarily selected Grover’s - type [40] quantum processors that could “measure” entangled proton qubit states in intervals, δt << 10-13 s [13]. Figure 5 illustrates approximate proton – electron lone-pair conFigureurations “seen by” Grover’s quantum readers in intervals, δt << 10-13 s. Table I illustrates model predictions of Grover’s processor’s measurements on entangled proton qubit states occupying G´-C´, *G-*C and *A-*T superpositions. Predictions in Table 1 agree with observation, except *A-*T sites (Figure 4) are deleted by measurements of Grover’s processor [16, 38]. Also, within subset intervals, Δt́ ≤ 10-14 s, entangled proton – quantum processors executed quantum information processing that included selection of electron lone-pairs and/or amino protons of accessible amino acids and classical incoming tautomers. This selection specified peptide bond formation [36, 57], and the ensuing time-dependent base substitution, ts, or time-dependent deletion, td, respectively [15, 16, 17, 37, 38, 39]. Ultimately, decoherence [13, 14], τD< 10-13 s, of proton – processor entanglements provided energy for peptide bond formation ~ 8 to 16 KJ/m [28] and caused the quantum mechanical superposition system to collapse [35, 36, 37, 38, 39] onto a selected classical isomer, which specified the time-dependent base substitution, ts, at this base pair site [15, 16, 17, 54]. The model implies Grover’s-type [40] quantum reader measurements of 20 accessible entangled proton qubit states within ancestral RNA – protein systems generated a triplet genetic code of 43 codons specified by ~ 22 L- amino acids [35, 36, 57]. When primitive duplex RNA genomes became too massive and unwieldy for acceptable “error-free” duplication [35, 36, 58, 59, 60, 61], rudimentary repair enzymes [139]were selected that replaced duplex RNA with a more stable DNA genome [28, 35, 36, 57]. The third phase, a DNA – protein system, was thus introduced, which also initiated the OoSL phase of evolution [57, 63]. A schematic of implied incremental increases in “genomic versatility” is illustrated in Figure 17. This model displayed in Figure 17 implies life’s origin emerged, and was sustained, in terms of approximate evolutionary increments [35, 36, 37, 38, 39]. A set of increment arrows (→) in Figure 17 identifies the following sequential (or simultaneous) evolutionary developments over the three phases of primordial evolution: monomers → oligomers → ribozymes → duplication of nucleotides → duplex RNA polymer segments → entangled proton qubits → ribozyme – proton entanglements → quantum transcription of entangled qubit states → quantum translation of entangled qubit message → quantum selection of triplet code → construct polypeptides → enzymes from ribozyme – proton entanglements → replication via enzymes → introduction of repair enzymes → genome chemistry selection, RNA replaced by DNA → duplex DNA genomes, etc. This OoSL model implies quantum entanglement algorithms, developed and implemented in ancestral RNA – protein systems, were subsequently retained and refined within evolving duplex DNA systems. Classical mechanisms do not explain evolutionary processes between ~ –4.1×109 y and ~ – 3.7×109 y [57, 63, 83], but EPR-entanglement algorithm processes [7, 8, 9, 10, 11] provide plausible mechanistic explanations [35, 36, 37, 38, 39]. This argument allows explanations for origins of quantum information processing algorithms exhibited by ancient [46] T4 phage DNA [15, 16, 17], rodent – human microsatellite DNA evolution [38, 52], and 21st- century human genomic systems [37, 39, 47, 48, 49, 50, 51, 54, 100, 104]. In this case, a reverse-time extrapolation — of proton quantum dynamics required for observable quantum information processing exhibited by ancient [46] T4 phage DNA [15, 16, 17] — appears applicable to analogous, metabolically inert [64], duplex RNA segments occupying primordial pools (0 0C to 20 0C, pH 7 [18, 19, 20, 21, 22, 36, 89]) of ribozyme ‒ RNA systems [57, 58, 59, 60, 61].
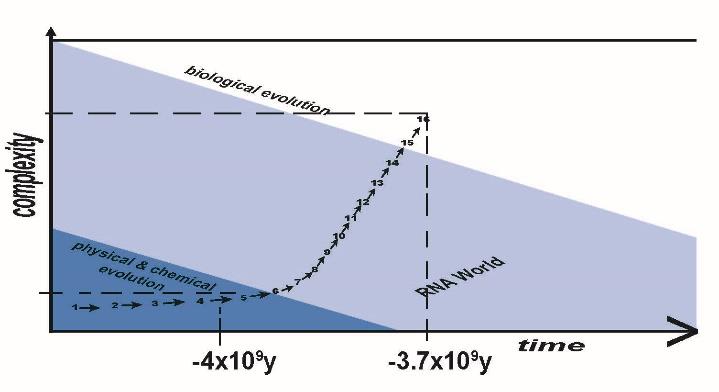
Figure 17: Incremental evolutionary transitions from RNA-ribozyme → RNA-protein → DNA-protein (Figure 17 Evolutionary increments during “classical” chemical – physical evolution (dark blue), and during entangled proton qubit-enabled RNA evolution (light blue), yielding duplex DNA systems of regular “biological evolution” (white background). Earliest life-forms are identified at – 4×109 y and earliest stromatolites [83] at – 3.7×109 y.) Based on ribozyme – RNA evidence [57, 58, 59, 60, 61, 82, 83, 84] and the model discussed here [35, 36], ancestral RNA genomic structures were susceptible to significant evolutionary pressures that allowed Darwinian selection to exploit “advantageous” applications of quantum information theory [40] involving (a) the creation [29, 30, 31] and measurement [35, 36, 37, 38, 39] of entangled proton informational qubits [41, 42, 43, 44, 45], (b) quantum/classical genomic informational interfaces [35, 36, 37, 38, 39] and (c) enzyme – proton entanglements that implement quantum searches, Δt′ ≤ 10_−14_ s [13], to specify synthesis information for a “new” base pair, evolutionary molecular clock event, ts [15, 16, 17, 35, 36, 37, 38, 39, 100]. Also, replacement of ribozyme function with protein enzymes implies peptide-ribozyme – proton entanglement processes selected relevant amino acids to construct the protein enzyme replacement of ribozymes. When ancestral RNA genomes became too massive for acceptable “error-free” duplication, rudimentary repair enzymes were invoked that selected DNA over duplex RNA [28, 137, 138, 139]. Although enzymatic quantum information processing provided a selective advantage for duplex RNA, as living RNA systems became more complex and versatile, the duplex RNA genome became too “massive” for acceptable error-free duplication [35, 36, 37, 57]. Consequently, rudimentary genome duplication “repair” systems were introduced that selected DNA over duplex RNA for “reduced error” genome duplication [139, 154, 155]. In this case, quantum processing information enzymes (QPIE) gradually expedited genomic evolution from (i) the era of pre-LUCA RNA “genome-like” polymers, (ii) to the “complete” duplex RNA genome, (iii) to the RNA-DNA reverse transcriptase genome, (iv) to double helical DNA genomes [57, 58, 59, 60, 61, 155]. These postulated four stages of pre-cellular genome evolution, and contents of their corresponding primordial pools, are schematically represented in Figure 18 (from Koonin et al. [61] with permission). This model implies a form of Grover’s-type [40] quantum information processing has been operational over the past ~ 3.6 or so billion y [35, 36, 37, 38, 39]. The enzyme quantum reader was refined during the developmental era of duplex RNA genomes [57, 58, 59, 60, 61], and has been retained and “fine-tuned” for analogous operations in double helical DNA systems [15, 16, 17, 35, 36, 37, 38, 39, 52, 53, 54, 100, 101, 102, 103, 104]. Consequently, accumulated entangled proton qubit states within G′-C′, *G-*C and *A-*T sites of modern double helical DNA are “transparent” to the recently evolved DNA repair system [139], but are recognized and processed by the “earlier evolved” RNA “repair” system [35, 36, 154], using enzyme-proton quantum entanglements to implement molecular clock, ts and td [15, 16, 17, 35, 36, 37, 38, 39, 54]. Soon after genome conversion from duplex RNA to double helical DNA [28, 57], rates of keto-amino → enol- imine arrangement (Figure 3) responsible for ts were reduced by ~ 50- to 100-fold, because of 5-methyluracil (thymine) replacing uracil and cytosine replacing 5HMC [46]. This quantum-based evolutionary selection provided a “favorable” ts:td ratio, exhibited as stochastic random genetic drift [100, 101, 102, 103], for double helical DNA. Table 5 data [17] imply rates ts (τ ≈ 3200 y) ≥ 1.5-fold td (τ ≈ 6000 y) which would generate a slight A-T richness, consistent with observation [28, 100] and model prediction [15, 16, 17, 35, 36, 37, 38, 39]. In this case, stochastic random genetic drift [103] is a consequence of EPR entanglement- enabled ts + td [35, 36, 37, 38, 39].
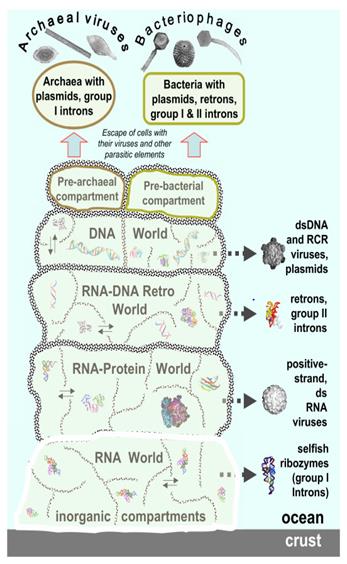
Figure 18: Evolution of genomic lineages from the primordial gene pool. ((from Figure 2, Koonin et al. [61] with permission). Characteristic images of RNA and protein structures are shown for each postulated stage of evolution, and characteristic virion images are shown for emerging classes of viruses. Thin arrows show the postulated movement of genetic pools between inorganic compartments. Block arrows show the origin of different classes of viruses at different stages of pre-cellular evolution.) A reverse-time extrapolation from observables [15, 16, 17, 18, 19, 20, 21] exhibited by ancient T4 phage DNA [46] implies that entangled proton qubits could have originally emerged in the first “susceptible” ancestral duplex segments of primitive RNA – ribozyme systems [58, 59, 60, 61]. This assumption requires primordial ribozyme – RNA duplex segments to simulate, approximately, conditions exhibited by ancient T4 phage DNA systems that accumulate EPR-generated entangled proton qubits in metabolically inert (extracellular [64], pH 7, 20 0C) base pair isomer superpositions, observed as G′-C′, *G-*C and *A-*T [15, 16, 17, 18, 19, 20, 21, 22, 23]. This scenario provides a possible source of “RNA-type” hydrogen bonded duplex molecules [28, 65] susceptible to occupancy by EPR-generated [29, 30, 31] entangled proton qubits [15, 16, 17, 35, 36, 37, 38, 39, 54, 104], and thus, allowed ancestral peptide-ribozyme – RNA systems [57, 58, 59, 60, 61] to form entanglement states with oscillating, │+>⇄ │–>, entangled proton qubits, occupying decoherence- free subspaces [11, 67, 68, 69] within hydrogen bonded [65] duplex RNA segments [35, 36]. At this stage of evolutionary development, peptide-ribozyme – proton entanglements could implement an entanglement- directed quantum search, Δt′ ≤ 10‒14 s < τD< 10−13 s [13], to select the next amino acid electron lone-pair, or amino proton, to be added to the pre-protein peptide polymer [35, 36]. Additionally before proton decoherence, τD< 10−13 s, operations of the entangled ribozyme – proton system included (a) generating a transcribed message based on quantum informational content of “measured” entangled proton qubits [15, 16, 17, 37, 38, 39], (b) implementing an entanglement-directed quantum search, Δt′ ≤ 10_−14_ s [13], that specifies the incoming base’s electron lone-pair, or amino proton, for evolutionary substitution, ts, or deletion, td, and (c) feedback responding “Yes” or “No” to translation of the transcribed “qubit message” [16, 17]. “Yes” implies existence of an “r+-_type” allele which allows replication initiation, but “No” identifies an unacceptable “mutant allele”, and therefore, replication is denied. Before “DNA-type” repair enzymes were introduced [139, 154, 155], RNA – ribozyme systems avoided evolutionary extinction by disallowing conserved, “_r+- type” allele contribution to the gene pool when “excessive” EPR-generated mutations (entangled qubits) were present [23, 35, 36]. Other “relaxed” genes were allowed mutation, variation and selection. These processes introduced viable peptide-ribozyme – RNA, wild-type “r+-systems” where peptide-ribozyme – proton entanglements were exploited to generate rudimentary peptide chains that subsequently usurped ribozyme functions. When duplex RNA genomes became “too
massive” for efficient, “error-free” duplication, “repair” enzymes [139] were selected that ultimately replaced RNA with DNA [154, 155], thereby introducing DNA – protein systems [35, 36]. Koonin’s [57] assessments imply nascent DNA – protein systems possess sufficient evolution potential to evolve into more complex living systems and organisms. In this case, Koonin’s Many Worlds in One (MWO) hypothesis [57] that the probability of existence of any possible evolutionary scenario in an infinite multiverse is exactly 1 is not required. Based on the scenario outlined here, if entangled proton qubits are not ignored as done in original studies of time-dependent evolution dynamics exhibited by (i) T4 phage DNA [18, 19, 20, 21, 22], (ii) human gene systems [35, 37, 38, 39, 53, 54, 104], and (iii) human – rodent microsatellites [38, 52] the MWO hypothesis is not required to explain origin and evolution of life on an “Earth-like” planet in Earth’s universe. Entangled proton qubit explanations [35, 36] allow life-forming polymers to originate in an ancestral RNA – ribozyme system [58, 59, 60, 61], where selected quantum bioprocessors [40] simulate a “truncated” Grover’s quantum search to “measure” entangled proton qubit states. This provides a hypothesis for origin of the triplet genetic code, utilizing 43 codons for ~ 22 L-amino acids [35, 36, 57]. Consequently, ribozyme-peptide “processing” of entangled proton qubits could generate RNA – protein systems where “repair” enzymes [139] ultimately intercede to replace unstable RNA with DNA. Subsequent quantum entanglement algorithmic processing of entangled proton qubits allows DNA – protein systems to further evolve on Earth as observed [57, 58, 59, 60], and originate and evolve on other “Earth-like” planets in Earth’s universe [35, 36].
Discussion and Conclusion
Quantum theoretical predictions subjected to appropriate experimental challenges have always confirmed predictions [1]. These experimental tests focused on relatively isolated quantum mechanical systems [2, 3], such as an electron, atoms, small molecules and near-perfect crystals, all of which are susceptible to measurements by physics laboratory techniques [1, 2, 3, 11]. Since observable reactive biological systems are generally assumed to be embedded in “wet and warm” in vivodecohering environments [5, 6], contributions by superpositions of entangled states [7, 8, 9, 10, 11] wereconsidered negligible in reactive biological systems [4].
However, recent studies [35, 36, 37, 38, 39] of normal in vivo duplex DNA, containing keto ─ amino (─NH2) hydrogen bonds,exhibitreactive processes of EPR-arrangements [29, 30, 31], keto-amino ― (entanglement) → enol−imine, due to hydrogen bonded amino protons encountering quantum uncertainty limits, Δx Δpx ≥ ħ/2 [15, 16, 17, 54, 66]. This introduces probabilities of direct quantum mechanical proton – proton interaction, yielding EPR- arrangements [29, 30, 31], observed as [15, 16, 17, 54] G-C → G´- C´, G-C → *G-*C and A-T → *A-*T. Reduced energy product enol and imine protons occupying heteroduplex heterozygote [15, 16, 17, 23] base pair sites G´-C´, *G-*C, *A-*T are consequences of EPR-generated [29, 30, 31], entangled proton qubits [35, 36, 37, 38, 39], shared between two different indistinguishable sets of electron lone-pairs belonging to decoherence-free subspaces [11, 67, 68, 69] of enol oxygen and imine nitrogen on opposite genome strands (Figure 1-4). Product enol-imine proton qubit-pairs contain quantum-enhanced genetic information [43, 44, 45], and participate in entangled quantum oscillations, │+>⇄│─>, at ~ 4×1013 s−1 (~ 4800m s−1) between near symmetric energy wells until measured, δt << 10−13 s, in a major or minor genome groove (~ 12 or 22 Å, ) [70], by evolutionarily selected Grover’s [40] quantum bio- processors. This measurement creates an entanglement state between measured “groove” protons [70] and the enzyme quantum processor [40], which yields ts + td after (i) an initial formation of enzyme-proton entanglement, δt << 10−13 s, (ii) implementation of an entanglement-assisted enzyme quantum search (Δt′ ≤ 10_−14_ s), (iii) specification of the “correct” complementary mispair (Figure 6), and (iv) selected replication-substitution or deletion [15, 16, 17, 35, 36, 37, 38, 39, 54], with classical tautomers containing decohered protons. Quantum processor measurements [40] of quantum informational content, occupying heteroduplex heterozygote [23] sites G´-C´, *G-*C, and *A-*T sites (entangled proton qubit states) specify time- dependent substitutions, ts, exhibited as G′2 0 2 → T, G′0 0 2 → C, *G0 2 00 → A& *C2 0 22 → T (see Table 1 & Figure 5 legend for notation) whereas, time-dependent entanglement-generated deletions, td [16, 17], are exhibited as *A → deletion and *T → deletion. These observables [15, 16, 17, 20, 21] are not compatible with classical [27, 28] transcription and replication, but are entirely consistent with Grover’s [40] enzyme quantum processors {see Equation (15)}, measuring quantum informational content [35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45] embodied within EPR- generated [29, 30, 31] ─ keto-amino ―(entanglement)→ enol−imine ─ entangled enol and imine proton qubit pairs.
(Here bold italics distinguish entanglement originated ts, e.g., G′ → T, from classical Newtonian substitutions, e.g_., G → T [27, 28]). Random stochastic genetic drift [101, 102, 103] is explained by EPR-generated _ts + td [15, 16, 17, 35, 36], where measured rates, ts ≥ 1.5-fold td, Table 5 [16, 17]. Entanglement-generated ts, e.g., G′2 0 2 → T, etc. [35, 36, 54], are mechanistically, and therefore, biologically distinguishable from classical “Muller-type” [71] base substitutions, e.g., Newtonian, G → T [28, 139]. When rII mutant [23, 73] T4 phage systems require substitutions G → T or C → T to express the r+ allele, and G′ and/or *C of Gʹ-Cʹ and/or *G-*Cheteroduplex heterozygote sites [23] are located on the transcribed strand [20, 21], growth conditions routinely exhibit identical G′ → T and *C → T mutation frequencies expressed by pre-replication transcription, and post- transcription replication [16, 17]. Curiously, entanglement-enabled biochemical genetic pathways responsible for EPR-generated mutations, ts and td, exhibited by ancient [46] T4 phage [15, 16, 17, 18, 19, 20, 21] are directly applicable to evolving microsatellites, STRs [52], of rat and human genomic systems [38]. This conclusion is based on the fact (Section 5) that evolutionary distributions of the 22 most abundant microsatellites, STRs (Table 3) [52], common to rat and human are predicted by the EPR-generated, quantum entanglement algorithm for describing, ts and td, and their consequences [37, 38, 54, 104], exhibited by ancient [46] T4 phage DNA systems. These results are displayed by recognizing the construction of two “ordered sets” of expanding, and contracting, STRs in Tables 4a & 4b, using observables from Table 3 [52]. Evolutionary distributions of the 22 most abundant microsatellites (Table 3) common to rat and human [52] are qualitatively predicted (Tables 4a&4b) by quantum entanglement algorithm analyses, in Section 5 [38]. When measured by Grover’s-type quantum processors, δt ≤ 10–
13 s, microsatellites whose EPR-generated entangled proton qubits introduce a preponderance of initiation codons UUG, CUG, AUG, GUG participate in the expansion mode of DNA synthesis, but if more stop codons UAA, UGA, UAG were introduced and/or the particular sequence consisted exclusively of A-T, such microsatellites would generally decrease in relative abundance over evolutionary times. This quantum information processing model [37, 38] accurately predicts evolutionary distributions of the 22 most abundant microsatellites, STRs [52], common to rat and human. The mouse-rat-human evolution era is separated by ~ 70×106 y [129]. Agreement between model predictions [36, 37, 38, 39] and observations [18, 19, 20, 21, 22, 52, 53, 104, 112] implies (a) that EPR-generated [29, 30, 31] entangled proton qubits [35, 36, 37, 38, 39] are “stable” occupying decoherence-free subspaces [67, 68, 69] until measured by Grover’s [40] quantum reader and (b) quantum information processing of EPR-generated entangled proton qubits has been operational in all sustainable, duplex genomic systems [35, 36, 37, 38, 39]. Agreement among EPR-generated ts and td data exhibited by ancient [46] T4 phage DNA [15, 16, 17, 18, 19, 20, 21], and evolving rat and human STRs [37, 38, 52, 54], implies Grover’s [40] quantum processors have been measuring quantum informational content of EPR-generated entangled proton qubits throughout the history of duplex DNA [28, 35, 36, 57, 58, 59, 60, 155, 156, 157, 158]. Results displayed in Tables 4a & 4b require years to decades of entangled proton qubit stability for quantum information processing of EPR-generated entangled proton qubits to be responsible for accurately specifying rat and human microsatellite evolutionary distributions [37, 52]. Consistent with stability requirementsand Figure 17, EPR-generated entangled proton qubits were initially introduced into susceptible duplex RNA–ribozyme segments [35, 36, 57, 58, 59, 60, 61]. In these cases, Grover’s-type [40] quantum processors were selected to repair and/or to process EPR-generated quantum-enhancedgenetic information occupying decoherence-free subspaces [11, 67, 68, 69] within ancestral RNA–ribozyme duplex segments. This implementation of quantum measurements and processing of EPR-generated entangled proton qubits, preferentially selected duplex RNA for RNA world (Figure 17) genomes [57, 58, 59, 60, 61]. When RNA genomes became too massive for acceptable, “error- free” duplication, repair enzymes [139] were invoked that preferentially selected DNA genomes [155] over RNA. Consequently, EPR-generated entangled proton qubits have been measured and processed in duplex DNA since its evolutionary existence [36, 57]. These identical lG′ → T and *C → T mutation frequencies expressed by pre-replication transcription and post-transcription replication [15, 16, 17, 20, 21, 38] are not explained by classical genetics [27, 28], but are entirely compatible with EPR-generated entangled proton qubits occupying G´-C´ and_*G-*C_ sites subsequently processed by Grover’s-type [40] quantum processors [16, 17, 36, 54]. The enzyme “quantum reader” initially measures entangled proton qubit states of G´ and *C (Figure 5), which immediately generates transcribed entangled qubit information, G′ → T and *C → T. Subsequently, entangled states, G′2 0 2 and *C2 0 22, are decohered, and these isomers are replicated as normal T22 0 22, thereby expressing identical mutation frequencies, $G' \rightarrow T$ and $*C \rightarrow T$, via transcription, and subsequently by replication [35, 36, 37, 38, 39].
Experimental and theoretical evidence implies that over the past ~3.7 or so billion years, pre-cellular [35, 58, 59, 60, 82, 83, 172], prokaryotic [15, 16, 17, 36, 156, 157] and eukaryotic [37, 38, 39, 57, 138] evolution had ample opportunity to select preferable, advantageous mechanisms for protecting the gene pool and CNGS [113, 114] against acquiring unsafe levels of entangled proton qubits in haploid and diploid genomes. Although DNA repair enzymes [139] were acquired during the transition from ancestral RNA to DNA genomes [28, 36, 154, 155], the originally selected quantum entanglement algorithm for RNA genomic evolution was retained, and further refined, for EPR-generated $ts$ and $td$ in DNA systems [35, 36, 37, 38, 39, 82, 100, 101, 102, 103, 104]. Since $ts$ and $td$ can introduce and eliminate initiation codons UUG, CUG, AUG, GUG and termination codons UAG, UGA, UAA susceptible STRs, e.g., $(CAG)_n$ ($n \geq 37$) [109], can exhibit “dynamic mutations” [37, 38, 104, 105]. In duplex DNA of human genomes, unstable repeats [37, 38, 53, 54, 104, 105, 106, 107, 108, 109, 110, 111] exhibit expansions and contractions via dynamic mutations [37] where $(CAG)_n$ sequences ($n > 36$) can exhibit expansions $\geq 10$ $(CAG)$ repeats in 20 y [53, 54, 109].
This observation implies the hypothesis that ancestral genomes implemented dynamic mutation expansions as consequences of specific $ts$ [15, 16, 17], introduced into susceptible STRs [37, 38, 52]. A “net” triplet repeat dynamic mutation [105] expansion rate of 13 repeats, e.g., $(CAG)_13 = 39$ bp, per 20 y for 3.5 billion y would generate a genome of $\sim 6.8 \times 10^9$ bp, which is “ballpark” compatible with bp content of the Homo sapiens’ genome [28, 109]. This entanglement-enabled “genome expansion” mechanism [37, 38] can account for genomic growth, over the past $\sim 3.5$ billion y, from primordial RNA, to $21^{st}$ century DNA of $\sim 6.8 \times 10^9$ base pairs [54, 104, 105, 106]. In this case, microsatellite content of prokaryotic and eukaryotic genomes would be proportional to genome size, which is consistent with observation [112].
Analogous to ancestral RNA genomes [35, 36, 60], modern “cancer genes” [55, 75, 140, 141, 142, 143], “Alzheimer’s genes” [76, 77, 144, 145, 146] and the huntingtin gene [53, 54, 109] containing unsafe levels of entangled proton qubits are disallowed further contribution to the gene pool, thereby serving as necessary “gatekeeper” genes that preserve a “wild-type” form of the human “gene pool” [35, 36, 37, 38, 39]. With exception of genes exhibiting unstable repeat diseases [53, 54, 104, 105, 106, 107, 108, 109, 110, 111], haploid “gatekeeper” genes that have acquired unsafe levels of EPR-proton qubits are eliminated during spermatogenesis or oogenesis, whereas unsafe diploid genes express age-related degenerative diseases [55, 56, 75, 76, 77] when entangled proton qubit acquisition exceeds an evolutionary selected “threshold limit” [35, 36, 37, 50]. This allows conserved noncoding genetic spaces (CNGS) [113, 114] to be preserved across the rat-mouse-human evolution era, $\sim 70 \times 10^6$ y [129].
Based on Section 5 analyses [38], entanglement-enabled information processing [7, 8, 9, 10, 11, 29, 30, 31, 37] cannot be simulated by classical models [40, 41, 42, 43, 44, 45, 52]. Accurate representations of microsatellite evolution requires inclusion of normal, EPR-generated [29, 30, 31] entangled proton qubits [16, 17, 52], which are subsequently processed by a Grover’s-type [40] quantum reader, thereby specifying observable evolution instructions with “measured” quantum information. Therefore, the time rate of change of biological noise, $dN(t)/dt$ [15, 16, 17, 101, 102, 103, 147, 148, 149, 150], must include quantum entanglement contributions, $\beta t$ (Appendix I), plus exclusively classical components, $\lambda$ [71], given in Eq. (17) as $dN/dt = \lambda + \beta t$. Equation (17) is integrated to obtain classical plus EPR-entanglement contributions, which are ultimately expressed as separate classical and entanglement contributions to age-related disease, given by Equation (22).
Analyses imply [35, 36, 37, 38, 39] EPR-entanglement terms, $\sum_j \beta_j t^4$ in Equation (22), are primary contributors to age-related disease. For example, time-dependent incidence of an age-related degenerative disease Huntington’s disease (Figure 14 [53]), cancer (Figure 15 [56]), Alzheimer’s disease (Figure 16 [77]) is approximated by measurements of quantum informational content within entanglement-enabled terms, $\sum_j \beta_j t^4$, in Equation (22); analyses indicate classical terms do not contribute to observable, disease incidence as a function of age data, exhibited in Figures 14-16. These analyses [35, 36, 37, 38, 39] imply that the relatively slow progress in understanding cancer’s origin and complexity [147, 148, 149, 150] can be attributed to “classical only” assessments, which previously neglected primary quantum entanglement contributions, identified by $\sum_j \beta_j t^4$ in Equation (22).
This conclusion implies an experimentally testable prediction regarding contributions by quantum information processing of EPR-generated entangled proton qubits when Huntington’s disease (Figure 14) is inherited, in terms of $(CAG)_{70}$ repeats [37, 54].
infants inherit an expanded (CAG)70 (n = 70 in Figure 14) Huntington’s disease genotype, phenotypic expression is delayed for ~2 to ~12 y after birth [53]. The EPR-entanglement Darwinian polynomial, Equation (22), concludes that the ~2 to ~12 y delay (Figure 14) after birth, before phenotypic expression, is due to (a) time required for EPR-proton qubits to populate the “threshold limit” [37, 49, 54] of the inherited (CAG)n sequence [64], after which (b) phenotypic expression is a direct consequence of Grover’s [40] quantum processors measuring quantum informational content of a “threshold limit” of entangled proton qubit states occupying CAG/GTC base pairs [37, 38]. The nonlinear graph in Figure 14 displays (CAG)n repeat length as a function of age-of-onset of Huntington’s disease [53], which is compatible with nonlinear contributions by $\sum_j \beta_j t^4$ terms in Equation (22).
Based on the present and other assessments [35, 37, 54], the time between birth and phenotypic manifestation of Huntington’s disease (Figure 14) is the time required for EPR-generated entangled proton qubits to populate the (CAG)n sequence to its “threshold limit”, and subsequently, exhibit phenotypic expression [35, 37, 54, 109] as consequences of quantum processors [40] “reading” quantum informational content embodied within EPR-generated entangled proton qubit states [35, 37]. The inherited (CAG)70 repeat establishes Huntington’s disease genotype [53, 109] that would be immediately expressed (hours, days, weeks) if standard Watson-Crick transcription and replication [28, 57] were implemented on keto-amino hydrogen bonded base pairs within the (CAG)70 repeat, which is contrary to fact (Figure 14). Observation [52, 53] and theory [35, 37, 38, 54] imply that phenotypic expression of Huntington’s disease requires Grover’s-type [40] quantum processors to measure quantum informational content in terms of EPR-generated entangled proton qubits, occupying a “threshold limit” [37, 109], within the inherited (CAG)70 repeat [37, 53, 54, 105, 106]. Other explanations are not obvious.
This and other reports [1, 35, 37, 38, 39, 54] imply the conclusion that delayed phenotypic expression of inherited (CAG)70 Huntington’s disease [53], and analogously, delayed expression of inherited (CTG)n (n ≥ 750) congenital myotonic dystrophy [111, 152], are due to phenotypic expression requiring the quantum information [42, 43, 44, 45] message generated by an initial quantum transcription [40] of EPR-generated entangled proton qubit states occupying a “threshold limit” of (CAG)n or (CTG)n repeat sequences. This quantum mechanical prediction [1] can be tested by comparing physical properties e.g., “stiffness”, flexibility, and melting temperatures of (CAG)n and (CTG)n sequences before and after an appropriate phenotypic expression [53, 111, 152]. Base pairs populated by EPR-generated entangled proton qubits, occupying decoherence-free subspaces [37, 38, 39, 68], between two different indistinguishable sets of electron lone-pairs, are bound more tightly, ~3 to ~7 Kcal/mole (Table 12), than metastable keto-amino hydrogen bonded base pairs [17, 65].
In cases of entangled proton qubits occupying adjacent base pairs within long repeat-sequences [109], the double helix would become less flexible and susceptible to “breakage”, exhibited by adjacent base pairs occupied by entangled proton qubits in (CCG)n repeats of Fragile X syndrome [104, 105, 106, 107, 108]. Also, the melting temperature of (CAG)n or (CTG)n repeats occupied by a “threshold limit” of entangled proton qubits should be detectably greater than those for “identical” (CAG)n or (CTG)n repeats occupied by metastable keto-amino hydrogen bonds at birth. Experimental confirmation of these quantum mechanical predictions would identify “new” accessible avenues for detecting, treating and/or preventing phenotypic expression of Huntington’s disease [53] and congenital myotonic dystrophy [111, 152], and other [106] unstable repeat human diseases.
Arguments presented here regarding manifestation of Huntington’s disease [37, 53, 109] and myotonic dystrophy [111, 152] via quantum measurements [37, 38, 39] of EPR-generated [29, 30, 31] entangled proton qubits — appear to be applicable in manifestation of ALS [78, 79]. Specifically, the model [37, 38, 39] implies phenotypic expression of ALS is, analogously, a consequence of quantumprocessor [40] measurements of EPR-generated entangled proton qubits occupying “threshold limits” [37] of expanded, G-C rich, hexanucleotide repeats, (GGGGCC)n [78, 79, 104, 111, 112, 113, 114].
The delay in recognizing quantum information processing [40, 41, 42, 43, 44, 45] of EPR-generated [29, 30, 31], entangled proton qubits [35, 36, 37, 38, 39] is due in part to “cover” provided by the molecular genetics history of observing [18, 19, 20, 21, 22, 23, 24, 25], but misidentifying, time-dependent EPR-generated — keto-amino — (entanglement) → enol-imine — entangled proton qubits [15, 16, 17] (Figures.1-4). Also, previous quantum physics models [5, 6] and quantum chemical consideration [163, 164, 165, 166, 167, 168, 169, 170] of Watson-Crick base pairs did not conclude that enol and imine hydrogen bonding states are stable. However, those investigations neglected unoccupied, lower energy enol and imine entangled proton qubit states that are subsequently populated by EPR arrangements [29, 30, 31], keto-amino → enol-imine, where entangled enol and imine proton qubit states are introduced [35, 36, 37, 38, 39]. Credible quantum molecular models must include accurate boundary conditions, consistent with observation [15, 16, 17, 18, 19, 20, 21]. In the case of EPR-generated [29, 30, 31] entangled proton qubits populating Gʹ-Cʹ, *G-*C and *A-*T sites [15, 16, 17, 35, 36, 37, 38, 39, 54], boundary conditions must account for quantum uncertainty limits, Δx Δpx ≥ ћ/2, operating on originally classical amino (−NH2) hydrogen bonded protons [65], which invoke probabilities of EPR arrangements [15, 16, 17, 29, 30, 31, 38], keto- amino → enol-imine, exhibited as time-dependent accumulations of heteroduplex heterozygotes, G-C → G′-C′ and G-C → *G-*C [15, 16, 17, 18, 19, 20, 21, 23]; *A-*T sites are deleted [16, 38]. Origination of heteroduplex heterozygotes [23, 123], and their transcription and replication properties [15, 16, 17, 18, 19, 20, 21], are not explained by classical models [27, 28], but are consistent with enzyme quantum reader-processor measurements [40, 41, 42, 43, 44, 45] of EPR-generated [29, 30, 31] accumulations [15, 16, 17, 18, 19, 20, 21, 22, 27] of intramolecular entangled proton qubit states [35, 36, 37, 38, 39], occupying decoherence-free subspaces [11, 67, 68, 69] of heteroduplex heterozygote,G′-C′ and *G-*C, superpositions [35, 36, 37, 38, 39, 54]. Prior quantum physics models [4, 5, 6] have implied in vivo environments of biological macromolecules are too “wet and warm” for significant biological contributions by quantum superpositions and entanglement states [7, 8, 9, 10, 11]. However, Darwinian selection has been operational for ~ 3.7 or so billion y [35, 36, 57, 58, 59, 60, 61, 83], and is executed at ambient biological temperature [37, 38, 39], and further, is not restricted to the macroscopic classical domain [4, 15, 16, 17, 18, 19, 20, 21, 35, 36, 37, 38, 39, 54]; so, existing quantum and classical laws of physics, chemistry and biology are available to participate in biological options on which natural selection operates [4, 28, 35, 36, 57, 82]. Necessary quantum mechanical processes exhibited by in vivo biological systems [15, 16, 17, 35, 36, 37, 38, 39, 171, 172] ─ e.g., photosynthesis [173, 174], avian navigation [4, 175], time-dependent genomic evolution [37, 38, 39, 100, 101, 102, 103, 104, 138] ─ are consequences of natural selection operating on available biological options for a relevant, “advantageous” biochemical function [176, 177]. Over evolutionary times, viable progeny were selected in terms of the more “advantageous” classical or quantum mechanical option [15, 16, 17, 35, 36, 37, 38, 39, 54, 100, 101, 102, 103, 104, 171, 172, 173, 174, 175], whereas deleterious options yield less robust progeny, and consequently, are generally eliminated by “purifying selection” [57, 176, 177]. Based on availability of “high resolution”, enzyme quantum reader measurements, δt << 10–13 s, of intramolecular entangled proton qubit states, │+>⇄ │−> [11, 35, 36, 37, 38, 39, 40, 54], theambient temperature, in vivo anti- entanglement hypothesis [4, 5, 6] is falsified. Since enzyme – proton entanglement reactions satisfy Δt′ ≤ 10−14 s [13, 35, 36, 37, 38, 39], ion incursions, H2O and random temperature fluctuations [12] do not obstruct evolutionarily selected enzyme – proton entanglement reactive processes, e.g., human-rodent microsatellite data (Tables 3, 4a-4b) [38, 52].Finally, consistent with Figure 14 and Sec. V, expansion and contraction of (CAG)n repeats [35, 37, 38, 39, 54, 109, 136] are explained by ts and td introducing and eliminating, termination and initiation codons. In this case, the “strand slippage” hypothesis [178] is not required [104]. Classical analyses [12] of reactive quantum phenomena [1, 2, 3, 4] generally yield nonsense [29, 30, 31, 32, 33, 34]. This and previous reports [35, 36, 37, 38, 39] identify several biological observables (phenomena) that require quantum theory for experimental tests ― e.g., (i) origin of molecular life-forms on planet Earth, (ii) evolutionary distributions of the 22 most abundant microsatellites common to rat and human, (iii) genotypic and phenotypic evolutionary dynamics exhibited by Huntington’s disease (CAG)n-repeats, (iv) origin of the triplet genetic code, utilizing 43 codons to specify ~ 22 L-amino acids, etc. ― but yield enigmatic confusion when analyzed classically [28, 52, 53, 57]. Evolutionary distributions of microsatellites common to rat and human genomes [52] are explained by quantum information processing of EPR- generated entangled proton qubits [38, 39]. In these cases, Grover’s-type [40] quantum processor “crawls” along major and minor genome grooves [70] at ~ 10–5 cm s–1 [115], where it “quantifies” quantum informational content of entangled proton qubit superpositions, │+> ⇄ │–>, oscillating between near symmetric energy wells at ~ 4´1013 s–1. In an interval, δt << 10–13 s, Grover’s processor “traps” an entangled-state proton, │–>, in a major or minor groove, which creates an enzyme – proton entanglement. Before proton decoherence ― Δtʹ ≤ 10–14 s < τD ― the enzyme – proton entanglement implements quantum information processing (Table 2), which specifies evolutionary distribution of rat and human microsatellites [37, 38, 39]. Analogous quantum information processing algorithms are routinely operational in DNA of human brain cells [179, 180], which are embedded within an evolutionarily designed neural circuitry [181]. Consequently, quantum information processing, Δtʹ ≤ 10– 14 s [13], executed by a single brain cell could communicate the resulting quantum information processing calculations to the brain’s neuronal network of ~ billons of neurons. Since empirical evidence [179] implies that consciousness [1, 180] could originate from specific computations, quantum information processing [35, 36, 37, 38, 39] by brain cell DNA [180] suggests a micro-physical “working hypothesis” for consciousness, governed by the neural circuitry network of quantum information processing DNA systems. Analogous to evolution instructions [35, 36, 37, 38, 39] provided by “measured” [40] quantum informational content embodied within EPR- generated entangled proton qubit-pairs [43, 44, 45], orchestrated quantum information processing of brain cell DNA [181, 182] could be responsible for the phenomenon of consciousness [1, 179, 180]. This and prior reports [35, 36, 37, 38, 39] conclude that quantum information processing [40, 41, 42, 43, 44, 45] of EPR-generated entangled proton qubit-pairs should not be neglected in reactive molecular genetic systems [e.g., 16,17,47-56,104,140-146], including quantum dynamics information processing of EPR-proton qubits in brain cell DNA [37, 38, 39].
Acknowledgement
Conservations with Roy Frieden on quantum coherence and decoherence of enzyme – proton entanglements in biological systems are gratefully appreciated. Comments on this research by Don Kouri and John Sabin were very helpful and are appreciated. I thank Jeremy Greiner for assistance in creating several Figure used in discussions of evolutionary distributions of human-rodent microsatellites. Faith Maina provided motivation and incentive to create the initial version of this report, for which I am sincerely thankful. An early version of this report was presented at the Texas Section of the American Physical Society Meeting, San Antonio, TX, 9-11 March 2017 (F2.0007), which was supported by the College of Education, Texas Tech University. The author states conflicts of interest do not exist.
References
-
Rosenblum B, Kuttner F (2011) Quantum Enigma: Physics encounters consciousness. 2nd (edn.) Oxford University Press, New York.
-
Merzbacher E (1997) Quantum Mechanics. 3rd (Edn.), John Wiley & Sons, INC New York.
-
Sakurai JJ, Napolitano JJ (2013) Modern Quantum Mechanics. 2nd (edn.), Pearson Educational India Limited, Upper Saddle River.
-
Moheni M, Omar Y, Engel GS, Plenio MB (2014) Quantum Effects in Biology. Cambridge University Press, Cambridge, UK.
-
Wiseman HM, Eisert J (2008) Nontrivial quantum effects in biology: A skeptical physicist's view. In: Abbott D, Davies PCW, Pati AK (eds), Quantum Aspects of Life, Imperial College Press, London, pp: 381-402.
-
Arndt M, Juffmann T, Vedral V (2009) Quantum physics meets biology. HFSP J 3(6): 386-400.
-
Horodecki R, Horodecki P, Horodecki M, Horodecki K (2009) Quantum entanglement Rev Mod Phys 81: 865-942.
-
De Vicente JI, Spee C, Kraus B (2013) Maximally entangled set of multipartite quantum states. Phys Rev Lett 111: 110502.
-
Pfaff W, Taminiau TH, Robledo L, Bemien H, Markham M, et al. (2013) Demonstration of entanglement-by- measurement of solid-state qubits. Nature Physics 9: 29-33.
-
Amico L, Fazio R, Osterloh A, Vedral V (2008) Entanglement in many body systems. Rev Mod Phys 80: 517-576.
-
Monz T, Kim K, Villar AS, Schindler P, Chwalla M, et al. (2009) Realization of universal ion-trap quantum computation with decoherence-free qubits. Phys Rev Lett 103(20): 200503.
-
Reif F (1965) Fundamentals of Statistical and Thermal Physics. McGraw Hill, New York.
-
Tegmark M (2000) The importance of quantum decoherence in brain processes. Phys Rev E 61: 4194- 4206.
-
Zurek WH (2009) Quantum Darwinism. Nat Phys 5: 181-188.
-
Cooper WG (1994) T4 phage evolution data in terms of a time dependent Topal Fresco mechanism. Biochem Genet 32(11-12): 383-395.
-
Cooper WG (2009) Necessity of quantum coherence to account for the spectrum of time-dependent mutations exhibited by bacteriophage T4. Biochem Genet 47(11-12): 392-410.
-
Cooper WG (2009) Evidence for transcriptase quantum processing implies entanglement and decoherence of superposition proton states. BioSystems 97(2): 73-89.
-
Drake JW (1966) Spontaneous mutations accumulating in bacteriophage T4 in the complete absence of DNA replication. Proc Natl Acad Sci USA 55(4): 738-743.
-
Drake JW, McGuire J (1967) Characteristics of mutations appearing spontaneously in extracellular particles of bacteriophage T4. Genetics 55(3): 387- 398.
-
Baltz RH, Bingham PM, Drake JW (1976) Heat mutagenesis in bacteriophage T4: The transition pathway. Proc Natl Acad Sci USA 73(4): 1269-1273.
-
Bingham PM, Baltz RH, Ripley LS, Drake JW (1976) Heat mutagenesis in bacteriophage T4: The transversion pathway. Proc Natl Acad Sci USA 73(11): 4159-4163.
-
Drake JW, Baltz RH (1976) The biochemistry of mutagenesis. Ann Rev Biochem 45: 11-37.
-
Ripley LS (1988) Estimation of in-vivo miscoding rates. Quantitative behavior of two classes of heat- induced DNA lesions. J Mol Biol 202(1): 17-34.
-
Kricker M, Drake JW (1990) Heat mutagenesis in bacteriophage T4: Another walk down the transversion pathway. J Bacteriol 172(6): 3037-3039.
-
Drake JW, Ripley LS (1994) Mutagenesis. In: JD Karam (Ed.), Molecular Biology of Bacteriophage T4, American Society for Microbiology, Washington DC, pp: 98-124.
-
Cooper WG (1993) Roles of evolution, quantum mechanics and point mutations in origins of cancer. Cancer Biochem Biophys 13(3): 147-170.
-
Drake JW, Charlesworth B, Charlesworth D, Crow JF (1998) Rates of spontaneous mutation. Genetics 148(4): 1667-1686.
-
Watson J, Baker T, Bell S, et al. (2013) Molecular Biology of the Gene. 7th (edn.), Menlo Park, CA: Benjamin-Cummings.
-
Einstein A, Podolsky B, Rosen N (1935) Can Quantum Mechanical Description of Physical Reality be Considered Complete?. Phy Rev 47: 777-780.
-
Schrödinger E, Born M (1935) Discussion of probability relations between separated systems. Mathematical Proceedings of the Cambridge Philosophical Society 31(4): 555-563.
-
Schrödinger E, Dirac PAM (1936) Probability relations between separated systems. Mathematical Proceedings of the Cambridge Philosophical Society 32(3): 446-452.
-
Bell JS (1964) On the Einstein-Podolsky-Rosen Paradox. Physics 1(3): 195-200.
-
Bell JS (1993) Speakable and Unspeakable in Quantum Mechanics. Cambridge University Press.
-
Bennett C, Wiesner SJ (1992) Communication via one- and two-particle operators on Einstein-Podolsky- Rosen states. Phys Rev Lett 69: 2881.
-
Cooper WG (2017) Origin of Life Insight: Reactive Transitions from Anthropic Causality to Biological Evolution. OMICS Group eBooks.
-
Cooper WG (2016) Quantum information processing model explains “early” and “recent” genome repair mechanisms. Res. & Reviews: Journal of Pure & Applied Physics.
-
Cooper WG (2016) Molecular dynamics responsible for observable Huntington’s disease (CAG)n repeat evolution. Ann Neurodegener Dis 1(2): 1009.
-
Cooper WG (2017) Evolution via EPR-entanglement algorithm execution. Journal of Biomedical Engineering & Medical Imaging.
-
Cooper WG (2018) Consequences of EPR–proton qubits populating DNA. Adv Quantum Chem 77.
-
Grover LK (1996) A fast quantum mechanical algorithm for database search. In: Proc. 28th Annual ACM Symposium on the Theory of Computing, ACM, Philadelphia, p: 212-219; Phys Rev Lett 1997, 79: 325.
-
Spiller TP, Munro WJ, Barrett SD, Kok P (2005) An Introduction to quantum information processing: Applications and realizations. Contemp. Phys 46(6): 407-436.
-
Zoller P, Beth T, Binosi D, Blatt R, Briegel H, et al. (2005) Quantum information processing and communication. European Physical Journal D 36(2): 203-228.
-
Vedral V (2006) Introduction to Quantum Information Science, Oxford University Press, UK.
-
Bub J (2010) Quantum Computation: Where Does the Speed-Up Come from?. In Bokulich A and Jaeger G (eds.), Philosophy of Quantum Information and Entanglement, Cambridge University Press, Cambridge, pp: 231-246.
-
Wilde MM (2013) Quantum Information Theory. Cambridge University Press, Cambridge.
-
Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, et al. (2003) Bacteriophage T4 genome. Microbiol Mol Biol Rev 67(1): 86-156.
-
Cooper WG (2010) Transcriptase measurement of coupled entangled protons yields new proton-enzyme quantum entanglement. In: Quantum Entanglement; AM Moran, Ed; Nova Scientific Publishers, Inc., Hauppauge, NY, pp: 1-35.
-
Cooper WG (2010) Evolutionarily designed quantum information processing of coherent states in prokaryotic and eukaryotic DNA systems. In: Computer Science Research and the Internet; Morris JE, (Ed.), Nova Scientific Publishers, Inc., Hauppauge, NY, pp: 1-43.
-
Cooper WG (2011) The molecular clock in terms of quantum information processing of coherent states, entanglement and replication of evolutionarily selected decohered isomers. Interdiscip. Sci 3(2): 91- 109.
-
Cooper WG (2011) Accuracy in biological information technology involves enzymatic quantum processing and entanglement of decohered isomers. Information 2(1): 166-194.
-
Cooper WG (2011) Quantum Darwinian evolution implies tumor origination.
-
Beckmann JW, Weber JL (1992) Survey of human and rat microsatellites. Genomics 12(4): 627-631.
-
Gusella JF, MacDonald ME, Ambrose CM, Duyao MP (1993) Molecular genetics of Huntington’s disease Arch Neurol 50(11): 1157-1163.
-
Cooper WG (2012) Coherent states as consequences of keto-amino → enol-imine hydrogen bond arrangements driven by quantum uncertainty limits on amino DNA protons. Int J Quantum Chem 112(10): 2301-2323.
-
Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, et al. (2010) Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci 107(43): 18545-18550.
-
Dix D, Cohen P, Flannery J (1980) On the role of aging in cancer incidence. J Theoret Biol 83:163-171.
-
Koonin EV (2012) The Logic of Chance. The Nature and Origin of Biological Evolution, Pearson, FT Press, Upper Saddle River, New Jersey.
-
Cech TR (2012) The RNA World in Context. Cold Spring Harbor Perspectives in Biology 4(7): a006742.
-
Gravette JV, Stoop M, Hud NV, Krishnamurthy R (2016) RNA–DNA Chimeras in the Context of an RNA World Transition to an RNA/DNA World. Angew Chem Int Ed 55(42): 13204-13209.
-
Shelke SA, Piccirilli JA (2014) Origins of life: RNA made its own mirror image. Nature 515(7527): 347- 348.
-
Koonin EV, Senkevich T, Dolja VV (2006) The ancient virus world and the evolution of cells. Biol Direct 1: 29.
-
Benner SA (2014) Paradoxes in the origin of life. Orig Life Evol Biosph 44(4): 339-343.
-
Scharf C, Virgo N, Cleaves HJ, Aono M, Aubert-Kato M, et al. (2015) Strategies for origins of life research. Astrobiol 15(12): 1031-1042.
-
Wootters WK, Zurek WH (1982) A single quantum state cannot be cloned. Nature 299: 802-803.
-
Scheiner S (1997) Hydrogen Bonding: A Theoretical Perspective. Oxford University Press, Oxford.
-
Kouri DJ (2014) Harmonic oscillators, Heisenberg’s uncertainty principle and simultaneous measurement precision for position and momentum.
-
Xu GF, Zhang J, Tong DM, Sjöqvist E, Kwek LC (2012) Nonadiabatic holonomic quantum computation in decoherence-free subspaces. Phys Rev Lett 109: 170501.
-
Xu G, Long G (2014) Universalnonadiabatic geometric gates in two-qubit decoherence-free subspaces. Sci Rep 4: 6814.
-
Suter D, Álvarez GA (2016) Protecting quantum information against environmental noise. Rev Mod Phys 88: 041001.
-
Wing R, Drew H, Takano T, Broka C, Tanaka S, et al. (1980) Crystal structure analysis of a complete turn of B-DNA. Nature 287(5784): 755-758.
-
Muller HJ (1950) Our load of mutations. Am J Hum Genet 2(2): 111-176.
-
Topal MD, Fresco JR (1976) Complementary base pairing and the origin of base substitutions. Nature 263(5575): 285-289.
-
Benzer S (1961) On the topography of the genetic fine structure. Proc Natl Acad Sci USA 47(3): 403-415.
-
Weber JL, Wong C (1993) Mutation of human short tandem repeats. Hum Mol Genet 2(8): 1123-1128.
-
Weinberg RA (2013) The Biology of Cancer, Garland Science, 2nd (Edn.), New York.
-
Cruchaga C, Haller G, Chakraverty S, Mayo K, Vallania FLM, et al. (2012) Rare variants in APP, PSEN1and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One 7(2): e31039.
-
Qui C, Kivipelto M, von Strauss E (2009) Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci 11(2): 111-128.
-
Dejesus Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, et al. (2011) Expanded GGGGCC hexanucleotide repeats in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72(2): 245-256.
-
Majounie E, Reton AE, Mok K, Dopper EG, Wait A, et al. (2012) Frequency of the C9ORF72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross- sectional study. Lancet Neurology 11(4): 323-330.
-
Bergin EA, Blake GA, Ciesla F, Hirschmann MM, Li J (2015) Tracing the ingredients for a habitable Earth from interstellar space through planet formation. Proc Natl Acad Sci USA 112(29): 8965-8970.
-
Li Y, Dasgupta R, Tsuno K (2015) The effects of sulfur, silicon, water, and oxygen fugacity on carbon solubility and partitioning in Fe-rich alloy and silicate melt systems at 3 GPa and 1600 0C; implications for core-mantle differentiation and degassing of magma oceans and reduced planetary mantels. Earth Planet Sci Lett 415: 54-66.
-
Hedges SB (2002) The origin and evolution of model organisms. Nat Rev Genet 3: 838-849.
-
Nutman AP, Bennett VC, Friend, CRL, Van Kranendonk MJ, Chivas AR (2016) Rapid emergence of life shown by discovery of 3,700-million-year-old microbial structures. Nature 537: 535-538.
-
Wolf YI, Koonin EV (2007) On the origin of the translation system and the genetic code in the RNA world by means of natural selection, exaptation, and subfunctionalization. Biol Direct 2: 14.
-
Weinberg S (2008) Cosmology. Oxford University Press, Oxford.
-
Hawking S (2014) The Illustrated, A Brief History of Time. The Universe in a Nutshell, Bantam Books, New York.
-
Ashtekar A, Corichi A, Singh P (2008) Robustness of key features of loop quantum cosmology. Phys Rev D 77: 024046.
-
Poplawski NJ (2012) Nonsingular, big-bounce cosmology from spinor-torsion coupling. Phys Rev D 85: 107502.
-
Goldman N, Tamblyn I (2013) Prebiotic chemistry within a simple impacting icy mixture. J Phys Chem A 117(24): 5124-5131.
-
Eschenmoser A (2007) The search for the chemistry of life’s origin. Tetrahedron 63: 12821-12844.
-
Engelhart AE, Hud NV (2010) Primitive Genetic Polymers. Cold Spring Harb Perspect Biol 2(12): a002196.
-
Callahan MP, Smith KE, Cleaves HJ, Ruzicka J, Stern JC, et al. (2011) Carbonaceous meteorites contain a wide range of nucleobases. Proc Natl Acad Sci U.S.A 108(34): 13995-13998.
-
Kwok S, Zhang Y (2011) Mixed aromatic–aliphatic organic nanoparticles as carriers of unidentified infrared emission features. Nature 479(7371): 80-83.
-
Hudgins DM, Bauschlicher CW Jr, Allamandola LJ (2005) Variations in peak position of the 6.2 µm interstellar emission feature: A tracer of N in the interstellar polycyclic aromatic hydrocarbon population. The Astrophysical Journal 632(1): 316- 332.
-
Gudipati MS, Rui Y (2012) In-situ Probing of Radiation-induced Processing of Organics in Astrophysical Ice Analogs—Novel Laser Desorption Laser Ionization Time-of-flight Mass Spectroscopic Studies. The Astrophysical Journal Letters 756(1): 24.
-
García-Hernández DA, Manchado A, García-Lario P, Stanghellini L, Villaver E, et al. (2010) Formation of Fullerenes in H-Containing Planetary Nebulae. The Astrophysical Journal Letters 724(1): 39-43.
-
Li Y, Dasgupta R, Tsuno K, Monteleone B, Shimizu N (2016) Carbon and sulfur budget of the silicate Earth explained by accretion of differentiated planetary embryos. Nature Geoscience 9: 781-785.
-
Feynman RP, Leighton RB, Sands M (1965) The Feynman Lectures on Physics. Vol. III, Addison- Wesley, New York.
-
Tegmark M, Wheeler JA (2003) 100 years of the Quantum. Sci Am 284: 68-75.
-
Hwang DG, Green P (2004) Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci USA 101(39): 13994- 14001.
-
Kumar S (2005) Molecular clocks: four decades of evolution. Nat Rev Genet 6(8): 654-662.
-
Bromham L, Penny D (2003) The modern molecular clock. Nat Rev Genet 4(3): 216-224.
-
Whitney KD, Garland T Jr (2010) Did genetic drift drive increases in genome complexity?. PLoS Genet 6(8): e1001080.
-
Cooper WG (1995) Evolutionary origin of expandable G C rich triplet repeat DNA sequences. Biochem Genet 33(5-6): 173-181.
-
Richards R (2001) Dynamic mutations: a decade of unstable expanded repeats in human genetic disease. Mol Hum Genet 10(20): 2187-2194.
-
Mirkin SM (2007) Expandable DNA repeats and human disease. Nature 447(7147): 932-934.
-
Fu YH, Kuhl DAP, Pizzuti A, Pieretti M, Sutcliffe J, et al. (1991) Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 67(6): 1047 1058.
-
Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM (2009) Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol 16(2): 226-228.
-
Semaka A, Creighton S, Warby S, Hayden MR (2006) Predictive testing for Huntington’s disease: interpretation and significance of intermediate alleles. Clin Genet 70(4): 283-294.
-
Pearson CE (2011) Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities!. PLoS Genet 7(3): e1002018.
-
Higham CF, Morales F, Cobbold CA, Haydon D, Monckton DG (2012) High levels of somatic DNA diversity at the myotonic dystrophy type 1locus are driven by ultra-frequent expansion and contraction mutations. Hum Mol Genet 21(11): 2450-2463.
-
Hancock JM (1996) Simple sequences and the expanding genome. BioEssays 18(5): 421-425.
-
Lujambio A, Portela A, Liz J, Melo SA, Rossi S, et al. (2010) CpG island hypermethylation-associated silencing of non-coding RNAs transcribed from ultraconserved regions in human cancer. Oncogene 29(48): 6390-6401.
-
Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, et al. (2011) Biological functions of microRNAs: a review. J Physiol Biochem 67(1): 129-139.
-
Davies PC (2004) Does quantum mechanics play a non-trivial role in life?. BioSystems 78(1-3): 69-79.
-
Goel A, Astumian RD, Herschbach D (2003) Tuning and switching a DNA polymerase motor with mechanical tension. Proc Natl Acad Sci USA 98: 8485-8491.
-
Wigner EP (1957) Relativistic invariance and quantum phenomena. Rev Mod Phys 29: 255-268.
-
Brinkmann B, Klintschar M, Neuhuber F, Huhne J, Rolf B (1998) Mutation Rate in Human Microsatellites: Influence of the Structure and Length of the Tandem Repeat. Am J Hum Genet 62(6): 1408–1415.
-
Love JM, Knight AM, McAleer MA, Todd JA (1990) Towards construction of a high-resolution map of the mouse genome using PCR analyzed microsatellites. Nucleic Acid Res 18(14): 4123- 4130.
-
Richard GF, Kerrest A, Dujon B (2008) Comparative genomics and molecular dynamics of DNA repeats in Eukaryotes. Micr Mol Bio Rev 72(4): 686-727.
-
Dib C, Fauré S, Fizames C, Samson D, Drouot N, et al. (1996) A comprehensive genetic map of the human genome based on 5264 microsatellites. Nature 380: 152-154.
-
Sainudiin R, Durrett RT, Aquadro CF, Nielsen R (2004) Microsatellite mutation models: Insights from a comparison of humans and chimpanzees. Genetics 168(1): 383-395.
-
Amos W (2016) Heterozygosity increases microsatellite mutation rate. Biol Lett 12(1): 20150929.
-
Amos W (2010) Mutation biases and mutation rate variation around very short human microsatellites revealed by human-chimpanzee-orangutan genomic sequence alignments. J Mol Evol 71(3): 192-201.
-
Rubinsztein DC, Amos W, Cooper G (1999) Microsatellite and trinucleotide-repeat evolution: evidence for mutational bias and different rates of evolution in different lineages. Phil Trans R Soc Lond B 354(1386): 1095-1099.
-
Nenguke T, Aladjem M, Gusella J, Wexler N, Arnheim N (2003) Candidate DNA replication initiation regions at human trinucleotide repeat disease loci. Hum Mol Genet 12(9): 1021-1028.
-
Sawaya SM, Lennon D, Buschiazzo E, Gemmell N, Minin V (2012) Measuring Microsatellite Conservation in Mammalian Evolution with a Phylogenetic Birth–Death Model. Genome Biol Evol 4(6): 636-647.
-
Varela MA, Amos W (2010) Heterogeneous distribution of SNPs in the human genome: Microsatellites as predictors of nucleotide diversity and divergence. Genomics 95(3): 151-159.
-
Kumar S, Hedges SB (1998) A molecular timescale for vertebrate evolution. Nature 392(6679): 917- 920.
-
Snowdon DA, Kane RL, Beeson WL, Burke GL, Sprafka JM, et al. (1989) Is early natural menopause a biologic marker of health and aging? Am J Public Health 79(6): 709-714.
-
Ehrenberg L, von Ehrenstein G, Hedgran A (1957) Gonad temperature and spontaneous mutation rate in man. Nature 180(4599): 1433-1434.
-
Torrellas G, Maciá E (2012) Twist-radial normal mode analysis in double-stranded DNA chains. Phys Lett A 376(45): 3407-3410.
-
Cooper WG (1979) Proton transitions in hydrogen bonds of DNA. A first order perturbation model Int. J Quantum Chem Quantum Biol Symp 16(6): 171- 188.
-
Chung M, Ranum L, Duvick L, Servadio A, Zoghb H, et al. (1993) Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I Nat Genet 5(3): 254-258.
-
Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, et al. (1994) Unstable expansion of CAG repeats in hereditary dentatorubral-pallidoluysian atrophy. Nat Genet 6(1): 9-13.
-
Zhang L, Leeflang EP, Yu J, Arnheim N (1994) Studying human mutations by sperm typing: instability of CAG trinucleotide repeats in human androgen receptor gene. Nature Genet 7(4): 531- 535.
-
Rosenberg SM (2001) Evolving responsively: adaptive mutations. Nat Rev Genet 2(7): 504-515.
-
Feng DF, Cho G, Doolittle RF (1997) Determining divergence times with a protein clock: Update and reevaluation. Proc Natl Acad Sci 94(24): 13028- 13033.
-
O’Brien PJ (2006) Catalytic promiscuity and the divergent evolution of DNA repair enzymes. Chem Rev 106(2): 720-752.
-
Illingworth CJR, Mustonen V (2011) Distinguishing driver and passenger mutations in an evolutionary history categorized by inference. Genetics 189(3): 989-1000.
-
Beerenwinkel N, Antal T, Dingli D, Traulsen A, Kinzler KW, et al. (2007) Genetic progression and the waiting time to cancer. PLoS Comput Bio 3(11): e225.
-
Anderson WF, Camargo MC, Fraumeni JF, Correa P, Rosenberg PS, et al. (2010) Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA 303(17): 1723-1728.
-
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, et al. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446(7132): 153- 158.
-
Sisodia SS, Tanz RE (2007) Eds. Alzheimer’s disease: Advances in Genetics, Molecular and Cellular Biology, Springer Science + Business Media, New York.
-
Pifer PM, Yates EA, Legleiter J (2011) Point mutations in Aβ result in the formation of distinct polymorph aggregates in the presence of lipid bilayers. PLos One 6(1): e16248.
-
Mosconi L, Rinne JO, Tsui WH, Berti V, Li Y, et al. (2010) Increased fibrillar amyloid-β burden in normal individuals with a family history of late- onset Alzheimer’s. Proc Natl Acad Sci USA 107(13): 5949-5954.
-
Little MP (2010) Cancer models, genomic instability and somatic cellular Darwinian evolution. Biol Direct 5: 19.
-
Foo J, Leder K, Michor F (2011) Stochastic dynamics of cancer initiation. Phys. Biol 8(1): 015002.
-
Feder T (2010) Physicists invited to apply their insights to cancer. Phys Today 63(5): 27-28.
-
Moore NM, Kuhn NZ, Hanlon SE, Lee JSH, Nagahara LA (2011) De-convoluting cancer’s complexity: using a ‘physical sciences lens’ to provide a different (clearer) perspective of cancer. Phys Biol 8(1): 010302.
-
Cooper WG (1996) Hypothesis on a causal link between EMF and an evolutionary class of cancer and spontaneous abortion. Cancer Biochem Biophys 15(3): 151-170.
-
Harper PS (2009) Myotonic Dystrophy, 2nd (Edn.), Oxford University Press, Oxford UK.
-
Driver JA, Lu KP (2010) Pin1: a new genetic link between Alzheimer’s disease, cancer and aging. Curr. Aging Sci 3(3): 158-165.
-
Domingo E, Escarmis C, Sevilla N, Moya A, Elena SF, et al. (1996) Basic concepts in RNA virus evolution. FASEB J 10(8): 859-864.
-
Takeuchi N, Hogeweg P, Koonin EV (2011) On the origin of DNA genomes: Evolution of the division of labor between template and catalyst in model replicator systems. PLoS Comput, Biol 7(3): e2002024.
-
Flugel RM (2010) The precellular scenario of genovirions. Virus Genes 40(2): 151-154.
-
Holmes EC (2011) What does virus evolution tell us about virus origins? J Virol 85(11): 5247-5251.
-
Koonin EV (2003) Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol 1(2): 127-136.
-
Cooper DN, Youssoufian H (1988) The CpG dinucleotide and human genetic diseases. Hum Genet 78(2): 151-155.
-
Orgel LE (1963) The maintenance of the accuracy of protein synthesis and its relevance to ageing. Proc Natl Acad Sci USA 49(4): 517-521.
-
Löwdin PO (1963) Proton tunneling in DNA and its biological implications. Rev Mod Phys 35: 724-732.
-
Löwdin PO (1965) Quantum genetics and the aperiodic solid: Some aspects on the biological problems of heredity, mutations, aging and tumors in view of the quantum theory of the DNA molecule. Adv Quantum Chem 2: 213-359.
-
Kryachko ES, Sabin JR (2003) Quantum chemical study of the hydrogen-bonded patterns in A-T base pairs of DNA: Origins of tautomeric mispairs, base flipping and Watson-Crick → Hoogsteen conversion. Int J Quantum Chem 91(6): 695-710.
-
Moser A, Guza R, Tretyakova N, York D (2009) Density functional study of the influence of C-5 cytosine substitution in base pairs with guanine. Theoret Chem Acc 122(3): 179-188.
-
Pérez A, Tuckerman ME, Hjalmarson HP, von Lilienfeld OA (2010) Enol Tautomers of Watson−Crick Base Pair Models Are Metastable Because of Nuclear Quantum Effects. J Am Chem Soc 132(33): 11510-11515.
-
Villani G (2010) Theoretical investigation of hydrogen atom transfer in the cytosine-guanine base pair and its coupling with electronic rearrangement. Concerted vs stepwise mechanism. J. Phys Chem B 114(29): 9653-9662.
-
Xiao S, Wang L, Liu Y, Lin X, Liang H (2012) Theoretical investigation of proton transfer mechanism in guanine-cytosine and adenine- thymine base pairs. J Chem Phys 137(19): 195101.
-
Brovarets OO, Hovorun DM (2014) Why the tautomerization of the GC Watson-Crick base pair via the DPT does not cause point mutations during DNA replication? QM and QTAIM comprehensive analysis. J Biomol Struct Dyn 32(9): 1474-99.
-
Brovarets OO, Zhurakivsky RO, Hovorun DM (2014) Is the DPT tautomerization of the long A•G Watson- Crick DNA base mispair a source of the adenine and guanine mutagenic tautomers?A QM and QTAIM response to the biologically important question. J Comput Chem 35(6): 451-66.
-
Godbeer AD, Al-Khalili JS, Stevenson PD (2015) Modelling proton tunneling in the Adenine – Thymine base pair. Physical Chemistry Chemical Physics 17: 13034-13044.
-
Lambert N, Chen YN, Cheng YC, Li CM, Chen GY (2013) Quantum biology. Nature Phys 9: 10-18.
-
Vattay G, Kauffman S, Niiranen S (2014) Quantum biology on the edge of quantum chaos. PLoS One 9(3): 89017.
-
Tamulis A, Grigalavicius M (2014) Quantum entanglement in photoactive prebiotic systems. Syst Synth Biol 8(2): 117-140.
-
Scholes GD, Mirkovic T, Turner DB, Fassioli F, Buchleitner A (2012) Solar light harvesting by energy transfer: from ecology to coherence. Energy and Environmental Science 5: 9374-9393.
-
Gauger EM, Rieper E, Morton JJL, Benjamin SC, Vedral V (2011) Sustained quantum coherence and entanglement in the avian compass. Phys Rev Lett 106(4): 040503.
-
Gillespie JH (2004) Population Genetics: A Concise Guide. The Johns Hopkins University Press.
-
Lynch M (2010) Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA 107(3): 961-968.
-
Petruska J, Hartenstine MJ, Goodman, MF (1998) Analysis of strand slippage in DNA polymerase expansions of CAG/CTG triplet repeats associated with neurodegenerative disease. J Biol Chem 273(9): 5204-5210.
-
Dehaene S, Lau H, Kouider S (2017) What is consciousness, and could machines have it? _Science_ 358: 486-492.
-
Stern P (2017) Neuroscience in search of concepts. Science 358: 464-465.
-
Donato F (2017) Assembling the brain from deep within. Science 358: 456-457.
-
Hameroff S, Penrose R (2014) Consciousness in the universe: a review of the ‘Orch OR’ theory. Physics of Life Reviews 11: 39-78.
-
Cooper WG, Kouri DJ (1972) N-particle noninteracting Green’s function. J Math Phys 13: 809-812.
-
Kadenbach B, Munscher C, Frank V, Muller Hocker J, Napiwotzki J (1995) Human aging is associated with stochastic somatic mutations of mitochondrial DNA. Mutation Res 338(1-6): 161-172.
-
Elango N, Kim SH, NICS Program, Vigoda E, Yi SV (2008) Mutations of different molecular origins exhibit contrasting patterns of regional substitution rate variation. PLoS Comput Biol 4(2): e1000015.
-
Walser JC, Furano AV (2010) The mutational spectrum of non-CpG DNA varies with CpG content. Genome Res 20(7): 875-882.
-
Zoete V, Meuwly M (2004) Double proton transfer in the isolated and DNA-embedded guanine- cytosine base pair. J Chem Phys 121(9): 4377-4388.
-
Nachman MW, Crowell SL (2000) Estimate of the mutation rate per nucleotide in humans. Genetics 156(1): 297-304.
-
Gurney RW, Condon EU (1929) Quantum Mechanics and Radioactive Disintegration. Phys Rev 33: 127- 140.
-
Zurek W (1991) Decoherence and the transition from quantum to classical. Phys Today 44: 36-44.
- Sense, Gravity, Parity & Chirality in Mathematical Physics
- Quantum Lattice Simulations PHYSICS: Microcircuit Particle Formation and Observable Macroscopic Irreversible Time - A Discrete Lagrangian with Cellular Automata Framework
- Quantum Biology from Biomacromolecule to Cell, and Central Dogma Described by Quantum Theory
- Focus, Agility, Speed and Technology (FAST) for Sustainability and Growth
- Square Root Metric Geometry and Pati-Salam Model in Curved Space-Time
- A Simple System Demonstrating the Mpemba Effect in Classical Mechanics