Possibilities of Viral Immune Evasion Exemplified by Human Papilloma Virus in HIV Infected People
Immune surveillance is of great significance for development of papillomavirus infections, as it has an impact on the disease course and its transformation into neoplasias. Immune response in infection of HIV patients with human papillomavirus deserves particular attention. Numerous studies suggest modification of immune response by the viruses in monoinfections (human immunodeficiency virus, human papillomavirus) and in combined infections. At present, numerous possible viral evasion mechanisms of innate and adaptive immunity factors are known. Despite a large body of accumulated knowledge on the HIV and papillomavirus infection course, early diagnosis and timely treatment of coinfected patients are hampered, which adversely affects their prognosis. There is still a need for expanding the techniques for early diagnosis of papillomavirus infection in HIV-infected individuals and searching for effective treatment methods.
Introduction
Human papillomavirus (HPV) presents a serious problem for public health worldwide. The infection that it causes has various manifestations from genital ad common warts to development of rectal, oropharyngeal, laryngeal carcinoma, carcinoma of the skin of the head and neck, cervical and vaginal carcinoma. Especially serious is the threat of neoplasia development in immunosuppressed patients, in HIV patients in particular. In women with HIV infection the risk of cervical carcinoma development is six times as high as that in women uninfected with HIV. High-risk oncogenic HPV types contribute seriously to development of skin and mucosa neoplasias.
Papillomavirus infection (PVI) is traditionally regarded as a sexually transmitted infection. The risk of HPV infection transmission through sexual contact is over 60%. In recent years, other transmission routes of the infection were proven: the intranatal one with injury of the oral epithelium, eye mucosa and genitals of the neonates. That is why timely HPV infection diagnosis in both adults and children presents an issue.
Human papilloma virus belongs to nonenveloped viruses: Monodnaviria domain, Shotokuvirae kingdom, Cossaviricota type, Papovaviricetis class, Zurhausenviraleos order, Papillomaviridae family [1]. The virus contains circular double-stranded DNA packed into an icosahedral capsid. HPV is an obligate epitheliotropic virus. HPV DNA replication occurs in the basal layer cells, the viral particlеs persist in the cells of other epidermal layers. In HPV infection, a low seroconversion rate and antibody production level are characteristic, in most cases, antibodies elicited after infection with one virus type, do not prevent infection with other HPV types [2].
By their ability to initiate neoplastic processes, papillomaviruses are classified into three groups: the oncogenic papillomaviruses of low (HPV 3, 6, 11, 13, 32, 34, 40, 41, 42, 43, 44, 51, 61, 72, 73); medium (HPV 30, 35, 45, 52, 53, 56, 58); and high oncogenic risk (HPV 16, 18, 31, 33, 39, 50, 59, 64, 68, 70) [3].
Currently, the main method of HPV prevention is vaccination. Three vaccines for primary prevention of the diseases associated with papillomavirus infection have been registered in the world: bivalent Cervarix® [4], tetravalent Gardasil® and nonavalent Gardasil-9® [5]. The vaccination previously proposed for female teenagers and women aged 9 to 26 years in 37 European countries has been turned to good advantage for PVI prevention in boys aged 9 to 15 years in a number of countries (Czechia, Austria, Germany, Italy, Croatia, Norway, Denmark and the UK). In the states whose population was immunized with HPV vaccine preparations, an immune layer of the population was formed. The vaccination coverage in some European countries reached over 70 %.
The only drawback of the current vaccines is impossibility to build immune protection against all the HPV types due to broad antigenic diversity of the pathogen. The current polyvalent vaccine provides the maximum protection against 9 HPV types (Gardasil 9). At present, the protective HPV antibody titer has not been determined, but the serologic studies established that the previous infection considerably reduces the risk of reinfection with HPV of the same type. Despite the use of vaccines against HPV, the vaccinated individuals are at risk of infection with other non- vaccine HPV types.
Beside vaccination, HPV prevention involves cervical screening aimed at early diagnosis and treatment of dysplasia as well as detection and treatment of cervical carcinoma. The current methods of cervical carcinoma diagnosis (cytological, histologic and molecular biological ones) have a number of drawbacks such as subjectivity and dependence of the result on the quality of the collected material, clinical pathologists’ expertise as well as impossibility to perform in laboratories that lack special equipment. In most cases papillomavirus infection is asymptomatic, so the patients do not always seek medical attention in due time.
As HPV infections remain common worldwide, their impact on the human population is considered rather serious. Due to the HPV body invasion patterns, no acute inflammatory reaction develops during the infectious process, and the virus is able to persist in the viral entry area as long as possible, thereby facilitating development of cervical, uterine, laryngeal, oropharyngeal and rectal carcinoma [6, 7].
For the sexually active population, the probability of contact with HPV is rather high, but this contact does not always lead to development of chronic HPV infection that can further progress to neoplasia [8].
It was shown that only 10-15% of the HPV-infected individuals retain a lifelong infection, as in most infected individuals at the early HPV infection stages a transient course of the infection with immunological clearance of the pathogen is possible [9].
Aspects of Papillomavirus Immune Evasion
HPV initially infects the basal cells of the cervical or rectal epithelium, of the skin, exploiting the properties of the late viral proteins L1 and L2. At the start of the infection process, early viral proteins E1-E8 facilitate virus replication and translation of the viral protein in the cells. In the epithelial cells, HPV proteins Е6 and Е7, interacting with cell cycle regulators p53 and pRb, block and decrease their activity. This inevitably leads to uncontrolled virus genome replication and chromosome instability of epithelial cells, their proliferation, dysplasia and malignization.
The process of HPV elimination from the body is largely a consequence of immune protection. At the early infection stage, the innate immunity factors contribute considerably to HPV recognition and elimination.
Mucous membranes and skin come first on the virus pathway. Damage to these barriers promotes HPV entry into the body. The presence of mucous protective proteins (such as mucin and lysozyme) on the surface also reduces the probability of HPV entry into the body [10].
Further, upon the virus entry into the host cell, the latter is recognized as a foreign one. Successful recognition and elimination of the viral pathogens at the early stage of HPV infection are facilitated by high expression of toll-like receptors: TLR2, TLR3, TLR7, TLR8 and TLR9. Interferon gene stimulator (STING) activation entails NF-Kb transcription factor and IRF3/IRF7 interferon-regulating factor activation.
It was shown that the cervical epithelium with low TLR9 expression is more sensitive to HPV, and the epithelial cells with high TLR9 expression are rarely infected with HPV [11]. It is also known that HPV protein E6 facilitates papillomavirus immune evasion by decreasing TLR9 expression and inhibiting TLR9 signaling cascade elements.
In patients with chronic viral infections, significant suppression of interferon synthesis is observed. Interferon production is the earliest reaction to the body’s contact with the virus, the interferon generates a protective barrier considerably earlier than the specific immune protection, stimulating the cell resistance. Upon receiving signals of foreignness, alpha- and beta-interferons hindering HPV entry and elimination of viral episomes from the damaged cells are produced [12]. The importance of this early non- specific response to the infection is undisputable.
Gamma-interferon is synthesized in activated Т helpers type 1 (Тh1) and natural killers (NK) and then in its turn activates macrophages. Interferon-gamma (IFN-γ) induces production of anti-inflammatory cytokines IL-1, IL-6, TNF-α and expression of МНС II antigens on the macrophages. Exposition with IFN-γ induces expression of Fc receptors for IgG that promotes phagocyte activity and antibody-mediated cytotoxicity. Therefore, interferons are critical factors in the antiviral immune response.
HPV E7 suppression of transcription of the genes responsible for TLR9 or IRF1, STING and interferon- stimulated genes (ISGs) leads to viral evasion of the immune response, impossibility to inhibit the virus-damaged cells and blockade of the apoptosis mechanism implementation.
Binding of interferon transcription regulatory factor (IRF-3) with HP16 protein E6 dampens IFN-β production. This as well proves the key position of IRF-3 in induction of the innate immune response against viral infections [13].
One of the early events in the contact with the papilloma virus is production of cytokines IL-1, TNF, IL-6 and IL- 8. Cytokine balance disorders can lead to impaired cell cooperation and immunity disbalance. In the early phase of non-specific immune response, the macrophages produce IL-12 and NK-IFN-γ. The key role in the activation of cell- mediated antiviral protection is played by anti-inflammatory IL-12 and other anti-inflammatory cytokines IL-1 and TNF that synergize with IL-12. The main aim of these cytokines is IFN-γ production by NK. The anti-inflammatory cytokine IL- 10 in its turn is a physiological inhibitor of IL-12 and IFN-γ synthesis. It is known that IL-10 facilitates differentiation of Т-naive cells to Т-regulatory (T-reg) ones. Which of the cytokines will prevail in immune protection activation to a large extent determines the choice of direction between the nonspecific protective reactions and adaptive immune response. Cytokines control the differentiation direction of СD4+ T lymphocyte that determine the form of specific immune response upon the infection entry. Differentiation to Th1 occurs in the presence of IL-12 and IFN-γ. Secretion of Th1, IL-2, IFN-γ and TNF promotes generation of specific cell immune response. Transformation of СD4+ T lymphocytes into type 2 Т helpers (Th2) is affected by IL-4. Th2 secrete IL-4, IL-5, IL-6 and trigger synthesis of specific immunoglobulins. The nuclear factor NF-κB plays an important role in transmission of the immune system signals. That is why this key factor of interferon and anti- inflammatory cytokine transcription becomes a target for papillomaviruses [14, 15]. The infection outcome and the possibility of viral persistence in the body largely depend on the balance of pro-anti-inflammatory cytokines.
Sensitive cell surface expression of the specific receptors which can consist of two or more receptor molecules is necessary for cytokine activation. It is noteworthy that both enhancement of cell cytokine production in the virus entry area and a sufficient number of receptors on the cells are necessary for effective cytokine system functioning.
For evasion of the immune response, proteins E6 and E7 inhibit signal transmission to the nuclear factor NF-кB, which leads to suppression of anti-inflammatory IFN-α, IL-6, IL-8 and TNF-α cytokine synthesis.
Protein E6 binds to IL-18, the main inductor of IFN-γ which leads to impairment of cell cytotoxicity. Blockade of IL-18-coding gene expression by Е6 and E7 viral proteins of suggests a mechanism of viral immune evasion.
A significant role in formation of inflammation belongs to chemokines. CXCL14 initiates direct chemotaxis of Langerhans cells (LC), dendritic cells (DC), NK and T cells into the HPV entry area [16, 17]. That is why for viral evasion of the immune response HPV modifies CXCL14 promotor methylation in a mediated manner through E7 to suppress the expression of the given chemokine. It is known that CCL2 and CCL20 chemokine expression is dampened by E6 and E7 proteins.
Chemokines CCL19 and CCL21 participate in directing DCs to lymphatic vessels. Similarly, expression of the related CCR7 receptor is necessary for presentation and initiation of DC specific immune response. Being exposed to CCL21, DCs move to larger vessels where they are passively transported by the lymph flow. Moreover, CCR7-mediated DC migration coordinates activation of specific T-regulatory (Т-reg) lymphocytes, thereby promoting peripheral immune tolerance [18].
DC are the key cells involved both in the innate and acquired immune response. For this very reason they are targets for viruses, in some cases being a reservoir of the pathogen. It is known that HPV minor protein L2 that hinders maturation of antigen-presenting cells promotes breakdown of intracellular transport of viral particles in DCs and LCs [19].
In response to emergence of HPV 16 virus-like particles (VLP), DCs secrete anti-inflammatory cytokines: IFN-α, IL-6, IL-8 and TNF-α. In HPV 16-infected keratinocytes, caspase-1 and IL-1 beta production is activated.
Presence of viral pathogens and virus-damaged cells, besides LCs and DCs, attracts NKs and Т lymphocytes with the NK (TNK) function to the damaged area [20].
NKs that lyse virus-attacked cells take an important part in papillomavirus elimination. For NK activation, sufficient expression of NKp30 and NKp45 receptors that decreases in HPV-16-associated intraepithelial lesions and cervical carcinoma is necessary. The fact of elevated HPV infection incidence rate and development of HPV-associated cancers in individuals with NK cell dysfunction has been proven [21].
However, the participation of the innate immunity components is not always sufficient for HPV elimination from the affected body, and then adaptive immunity factors get engaged in the virus elimination process. A particular role in virus elimination is given to Т-cytotoxic (CD8+) and Т helper cells (CD4+), Т-regulatory lymphocytes and the immune response that they mediate. The function of CD8+ и CD4+ Т cells both in the papilloma formation and in HPV elimination was experimentally proven on the murine papilloma virus model (MmuPV1) [22, 23].
Upon the entry of the viral antigens into antigen- presenting cells (DCs, LCs) antigen presentation through MHC class I and II occurs. This leads to generation of cytotoxic CD8+ Т cells of CD4+ helper Т-cell reactions, respectively. To present the epitopes of HPV viral proteins with participation of МНС class I stimulating T cells CD8+, the proteins are pre-processed and cleaved into smaller peptides by the proteasome of the antigen-presenting cells. The success of cell-mediated immune response in elimination of human papilloma virus-damaged cells largely depends on presentation of the viral epitopes by MHC I molecules. To evade recognition and immune response activation, papillomaviruses, as other viruses, suppress surface expression of MHC I molecules [24]. Е5 protein of bovine papillomavirus blocks MHC I transfer on the host cell surface, binding to several transmembrane cell proteins [25]. HPV 16 E7 suppression of MHC I molecules that leads to a decline in the activity of T-cytotoxic (СD8+) cells and a suppressive effect of HPV 16 protein E5 on the superficial expression of MHC II molecules and of СD1d that inhibits CD4+ T helper cell reactions and TNK response to the virus has been proven [26, 27]. HPV 16 E5 binds to the heavy α-chain of MHC I molecule with its hydrophobic region and so hinders its exit from the endoplasmic reticulum onto the cell surface [28].
For evading the immune response, viral protein E7 of HPV 16 causes immunosuppression of LC and CD8+ Т cells which is probably associated with the T-reg influx and cytotoxic Т cell tolerance.
Interesting is the role of the HPV L 1 and L 2 minor proteins. It is supposed that HPV 16 protein L2 blocks DC maturation, thereby disturbing the viral antigen presentation, and, subsequently, no cytotoxic Т-cell response activation occurs. Several HPV16 variants isolated from dysplastic and malignized epithelial cervical cells were detected, the structure of whose L1 and L2 proteins promotes breakdown of the viral capsid assembly. As a result of these changes, В-lymphocytes become unable to produce complete neutralizing antibodies to such mutated virions, and so facilitate evasion from the immune surveillance and persistence of poorly immunogenic viruses in the body [29].
For effective elimination of the HPV-associated infection, coordinated work of both innate and adaptive immunity is of key importance. Defects in some components of the immune response can lead to long-term virus persistence and oncogenic cell damage. Besides, getting an insight into the mechanisms of the HPV immune evasion mechanisms is significant for successful treatment and control of the viral infection.
Combined HPV Infection in HIV-Infected Individuals
There is no specific treatment against HPV, so the human immune system plays a critical role, especially in combined HIV and HPV infection.
Numerous publications are dedicated to studies of HIV infection; some of them deal with the immune status of HIV patients. In the recent years, a lot of effort has been made to improve HIV prognosis and prolong the patients’ survival. But, despite the antiretroviral therapy (HAART), one of the poor outcomes of the disease is development of neoplastic processes, including HPV-associated ones in HIV patients.
Synergism between HPV and HIV has not been studied to the full extent. It is supposed that HIV infection can promote damage of the epithelium that increases the probability of HPV acquisition [30].
In HPV-HIV coinfection, the immune response to HPV in HIV-infected individuals is modified. Due to the advances in medicine, HIV has been increasingly regarded as a chronic disease which can be controlled. However, HIV infection and the resulting chronic inflammation increase the concentration of the inflammatory markers, which leads to chronic immune activation and CD8+Т cell exhaustion. Besides, the CD4+ Т lymphocyte count decreases even in the patients on antiretroviral therapy. Interrelation between decreased СD8+ Т- lymphocyte activity and development of carcinomata has been proven. Combined chronic inflammation caused by HPV and HIV leads to cell exhaustion. Decreased CD4+ and CD8+ lymphocyte counts promote a decrease in the possibility of HIV and HPV clearance as well as development of dysplasia and progression of malignant tumors.
Thus, the outcome of HPV coinfection in HIV-infected individuals is determined by several factors:
- initial immunosuppression in the HIV-infected patients
- immunosuppression caused by papillomavirus invasion and the ability of the virus to evade immune response
- delayed HP diagnosis in HIV-infected individuals.
So the researchers have to face issues with the immunity features of the HIV-infected patients, a weak immune response to HPV entry and with search for new papillomavirus acquisition predictors in such patients.
Modification of Immune Surveillance of Papillomavirus Infections in HIV-Infected Patients
Patients with HIV-HPV coinfection have an elevated risk of precancerous cervical and anal canal, penile, oropharyngeal and vulvar lesions. The risk of carcinoma development is possible even in HIV-infected individuals with a normal CD4+ cell count [31, 32, 33]. Numerous cell protection defects in AIDS are associated with the ability of HIV to inhibit IL-12 synthesis. If production of the inflammatory cytokines (IL-1, TNF) is retained, IL-12 inhibition leads to long-term persistence of the pathogen in the host. Prognosis of the infection course is directly linked to the ability of the pathogen to stimulate IL- 12 synthesis. IL-18 plays a significant role in the HIV course. This anti-inflammatory cytokine is induced in response to IFN-α and β induction and facilitates IFN-γ production by natural killers. IL-18 is a strong chemoattractant for DC.
An elevated risk of neoplasia development in HIV-HPV coinfection is caused by multiple factors. Due to improvement in the HIV patient care their survival has been prolonged, and, as a consequence, the incidence of HIV-associated malignant neoplasia has increased [28, 29]. One of the main factors increasing the risk of neoplasia is HIV-associated immunosuppression in which the possibility of HPV-infected cell elimination is decreased. A low CD4+ Т cell count before HAART initiation is linked to a higher relapse risk in HIV- infected patients with HPV-associated anal squamous cell carcinoma (SCC) [34].
In response to HIV invasion, toll-like receptors (TLR7, TLR9) are expressed on DCs, and the regulatory factor interferon IRF7 is activated, with the result that plasmatic DCs produce type 1 interferons (α and β). DC are an important link between the innate and adaptive immunity. In chronic HIV infection, the plasmatic DC count in the peripheral blood decreases and, subsequently, IFN-α and β synthesis declines, which directly correlates to decreased CD4+ Т lymphocyte count and has a reverse correlation with the viral load and is associated with development of opportunistic infections. During chronization of the process in HIV-infected patients, DCs decrease the ability to produce IFN-α and β as a weak response to restimulation with TLR9 virus.
HPV-associated lesions have a higher content of CD4+ Т cells, DCs and macrophages which are known as targets for HIV infection, which makes HIV acquisition in these areas possible [35, 36].
It was previously thought that HIV mainly enters the cells via the surface CD4 receptors, but recent studies show that HIV employs additional receptors, including CXCR4 and CCR5 [37]. As is known, CXCR4, along with its ligand CXCL12, is involved in cell migration and chemotaxis [38]. CCR5 is also expressed mainly on leukocytes and is involved in several immunomodulation pathways [39]. Both receptors facilitate HIV entry into the cells by binding to the HIV envelope glycoprotein, which promotes the virus entry. After the entry into the cells, expression and replication of the HIV viral genes occurs, especially due to the activity of HIV Tat protein [40]. Therefore, epithelial cells are not only sensitive to HPV lesions, but are also subject to the impact of the expressing CD4, CXCR4 and CCR5 immune cells carrying HIV. HPV-HIV coinfection increases the HPV oncogenicity as the result of the changes in the immune system and the decline in the immune response to HPV caused by HIV [41]. Engagement of HIV Tat protein is suggested in the increase of the HPV oncogenic risk: the presence of HIV Tat not only enhances HIV gene expression, but it can also increase the expression of the genes of another viral DNA in the cells with productive intracellular levels of HIV Tat. This protein is integral to viral genome replication. HIV Tat binds RNA polymerase II and other proteins necessary for transcription, thereby facilitating viral DNA transcription and virus replication [42]. The role of HIV Tat in the HPV- associated oncogenesis was determined; Tat can increase the expression of Е6 and Е7 oncoproteins as well as that of Е2 protein which is responsible for HPV genome replication [43]. This interaction was exemplified by HPV-associated squamous epithelial cell transformation in the mucosa, anogenital and upper respiratory tract. In another study, Tat not only increased the level of HPV-associated oncogenesis, but also reduced p53 protein level, thereby increasing the malignancy potential [44, 45].
HIV coinfection alters cell-mediated immune response to HIV lesions, initially leading to CD4+ Т cell dysfunction; then HIV can alter the CD8+ Т lymphocyte function which is key for the immune response in HIV-associated lesions. In HPV infection, CD8+ Т cell immune response level is a significant predictor of the lesion progression and the future cancer risk [46, 47, 48]. HPV-infected cells are attacked by CD8+ Т cells, because E6 and E7 oncoproteins are processed and presented to MHC I. That is why E6- or E7-specific CD8+ Т cells can induce cytolysis of HPV-infected cells by way of perforin and granzyme B secretion. The anti-tumor effect of CD8+ Т cells is similar (Figure 1).
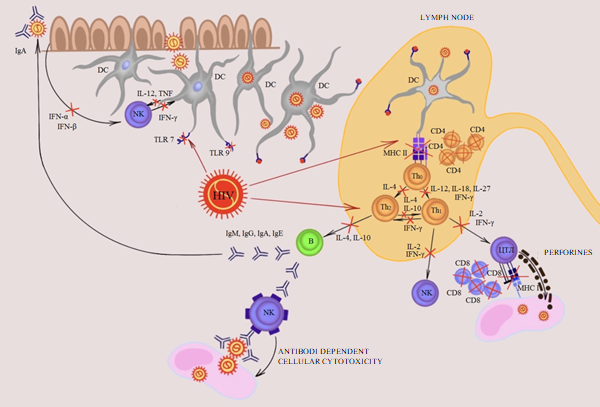
Receptor,
Activation,
$$ \longrightarrow $$ $$ \rightarrow $$
In patients with HIV-HPV coinfection, HIV infection facilitates FoxP3 overexpression in regulatory Т cells and leads to the local DC exhaustion [49]. Also, HIV can increase PD-L1 expression on CDs, thereby lowering their efficacy in the immune response to HPV and giving an additional emphasis to interaction between HIV and PD-1/PD-L1 [50].
As a study showed, in the women on antiretroviral therapy HIV decreased the DC density in the anal mucous membrane as part of HPV-induced immune response [51].
The ongoing inflammation and active immune response, especially IFN-γ secretion, cause elevated PD-L1 expression and thus activate PD-1/PD-L1 interaction. In terms of physiology, it serves as an important test of potentially overactive immune response and prevents immunopathology. PD-1 is an inhibiting receptor expressed on T cells; it controls Immunoglobulin, Inhibition Т cell function and proliferation by interaction with PD-L1 which can be expressed by regulatory Т cells, myeloid cells and tumor cells. HPV Е6 and Е7 oncoproteins increase PD-L1 expression, activating PD-1 protein reception and decreasing the local immune response to HPV-infected cells [52].
Active CD8+ Т- lymphocytes express PD-1 that physiologically serves as a balancing mechanism for inhibition of active immune reactions. In the context of CD8+ Т cell anti-tumor responses, as described above, tumors expressing PD-L1 can evade CD8+ Т cell cytotoxicity [53].
CD4+ Т cells play a critical role in maintaining CD8+ Т cell-mediated immune response HIV-HPV coinfection modifies the immune response, as HIV dampens the reaction to HIV-associated precancerous lesions, reducing the number of circulating CD4+Т-lymphocytes in patients whose HIV load is not controlled [54]. So it was shown that uncontrolled HIV infection reduces the anti-tumor activity of CD8+ T lymphocytes and, respectively, facilitates progression of precancerous lesions to malignant tumors.
It was also shown that HIV directly reduces the activity and efficacy of CD8+ Т lymphocytes. HIV infection causes chronic inflammation throughout the body associated with elevated inflammatory markers and chronic immune activation, and, in particular, with chronic CD8+ Т cell activation [55]. This condition of chronic immune activation leads to exhaustion of the CD8+ Т lymphocyte pool [56]. CD8+ Т cell exhaustion is considered to be partially mediated by the modulating effect of HIV on PD-1/PD-L1 expression; chronic activation of CD8+ Т lymphocytes leads to elevated PD-1 expression [57]. In HIV infection, interaction of PD-1 protein and PD-L1 ligand is getting increasingly significant, as the number of different cell types expressing this ligand increases. The above discussed PD-L1 expression on tumor cells is usually linked to poor prognosis, even in patients on HAART [58]. The risk of CD8+ Т cell exhaustion increases in HIV-HPV coinfection, combined chronic inflammation caused by both viruses also leads to exhaustion in patients on HAART. This reduces HPV clearance in the infected cells, which facilitates development of dysplastic lesions and progression to malignancy [59].
HIV infection leads to chronic inflammatory changes in the entire body. The progression of the infectious process increases PD-1 expression on CD8+ Т cells, reducing the activity of the systemic immune response. As CD8+ Т cells infiltrate dysplastic and malignant lesions and are critical for the anti-tumor response; this elevated PD-1 expression (secondary to chronic HIV infection) enables the PD-L1- expressing tumors to evade the anti-tumor response in HIV- infected patients.
Current Possibilities of Papillomavirus Infection Course Correction
Various methods of papillomavirus infection control are aimed at: the disease prevention (preventive vaccination), elimination of the lesion foci by surgical and cryodestruction methods (in local infection). But these methods are not always sufficient to eliminate the infection, and if this is the case, immunomodulators and the drugs that affect replication and transcription of the viral DNA get engaged in HPV control. One of the potential treatment methods is use of therapeutic vaccines. As distinct from preventive vaccines that are aimed at generating neutralizing antibodies to viral particles, therapeutic vaccines are intended to stimulate cell- mediated immune reactions for specific targeting and killing the infected cells. In numerous cases when HPV-associated lesions progress to carcinomata, HPV viral DNA will be integrated into a gene. As a rule, the integration process leads to deletion of numerous early (E1, E2, E4 and E5) and late (L1 and L2) genes. L1 and L2 gene deletion during HPV DNA integration makes preventive vaccines ineffective in terms of targeting the infected cells. Besides, Е2 is a negative regulator of HPV Е6 and Е7 oncogenes; so, E2 gene deletion during the integration leads to increased expression of these oncoproteins. It is supposed that this process promotes carcinogenesis of HPV-associated lesions, and uncontrolled E6 and E7 expression is considered a distinctive biological feature of HPV-associated carcinoma. As HPV Е6 and Е7 oncoproteins are necessary for generation and maintenance of HIV-associated malignant neoplasia, they are constantly expressed and stay transcriptionally active in transformed cells in HPV-induced carcinoma and precancerous lesions. Besides, as E6 and E7 are foreign proteins, they are able to circumvent a decrease in the immune tolerance to their own antigens, a problem associated with many other cancer types. So E6 and E7 are ideal targets for therapeutic vaccines against HPV [60]. These results motivated lots of effort on creating the optimal immunotherapy of HPV infections and diseases.
Most therapeutic vaccines contain E6 and E7 antigens in various forms and are aimed at delivering these antigens to antigen-presenting cells (APC) for antigen presentation stimulation through MHC class I and MHC class II. This leads to generation of cytotoxic CD8+ Т cells or CD4 + Т helper cell reactions, respectively. Before E6 and E7 antigens can be presented on the MHC class I molecule for stimulation of CD8+ Т-cell reactions, they are processed and digested into smaller peptides by the proteasome in APC. Not all the peptide fragments can be successfully loaded into the MHC molecule and recognized by antigen-specific T cells. Only some short peptides that contain a sequence of antigenic epitopes can bind to an MHC molecule with high affinity and interact with the receptor (TCR) of antigen-specific Т cells for gaining an immune response. Most therapeutic vaccines are focused on provoking immune reactions against E7, as it possesses better immunologic characteristics than E6 in preclinical models.
Conclusion
Despite the advances in prevention of HPV infections and HPV-associated malignant neoplasia, there is still a necessity to expand the methods of specific diagnosis of early infection stages and treatment of the current HPV infections; this problem is essential for HIV-infected persons.
There is a need for further studies of HIV impact on the immune response in HIV-HPV coinfection and a study of intercellular interactions of DCs, NKs, CD8+ and Т-reg (Foxp3) that promote precancerous and neoplastic lesions.
References
-
Lefkowitz EJ, Dempsey DM, Hendrickson RC, Orton RJ, Siddell SG, et al. (2018) Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res 46(D1): 708-717.
-
Bernard HU, Burk RD, Chen Z, van Doorslaer K, Hausen H, et al. (2010) Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401(1): 70-79.
-
de Villiers EM, Fauquet C, Broker TR, Bernard HU, Hausen HZ (2004) Classification of papillomaviruses. Virology 324(1): 17-27.
-
Szarewski A (2012) Cervarix®: a bivalent vaccine against HPV types 16 and 18, with cross-protection against other high-risk HPV types. Expert Rev Vaccines 11(6): 645-657.
-
Zhai L, Tumban T (2016) Gardasil-9®: A global survey of projected efficacy. Antiviral Research 130: 101-109.
-
Siegel RL, Miller KD, Jemal A (2017) Cancer statistics. CA: A Cancer Jourlan for Clinicans 67(1): 7-30.
-
Serrano B, Brotons M, Bosch FX, Bruni L (2018) Epidemiology and burden of HPV-related disease. Best Practice & Research Clinical Obstetrics & Gynaecology 47: 14-26.
-
Viarisio D, Gissmann L, Tommasino M (2017) Human papillomaviruses and carcinogenesis: well-established and novel models. Curr Opin Virol 26: 56-62.
-
Brianti P, De Flammineis E, Mercuri SR (2017) Review of HPV-related diseases and cancers. New Microbiologica 40(2): 80-85.
-
Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC (2013) Human papillomavirus and cervical cancer. Lancet 382(9895): 889-899.
-
Daud II, Scott ME, Ma Y, Shiboski S, Farhat S, et al. (2011) Association between toll-like receptor expression and human papillomavirus type 16 persistence. Int J Cancer 128(4): 879-886.
-
Joseph AW, Cody JW, Dohun P (2017) Evasion of host immune defenses by human papillomavirus. Virus Res 231: 21-33.
-
Haller O, Kochs G, Weber F (2006) The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344(1): 119-130.
-
Tummers B, Goedemans R, Pelascini LP, Jordanova ES, van Esch EM, et al. (2015) The interferon- related developmental regulator 1 is used by human papillomavirus to suppress NFκB activation. Nat Commun 6: 6537.
-
Richards KH, Doble R, Wasson CW, Haider M, Blair GE, et al. (2014) Human papillomavirus E7 oncoprotein increases production of the anti-inflammatory interleukin-18 binding protein in keratinocytes. Journal of Virology 88(8): 4173-4179.
-
Shurin GV, Ferri RL, Tourkova IL, Perez L, Lokshin A, et al. (2005) Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol 174(9): 5490-5498.
-
Cicchini L, Westrich JA, Xu T, Vermeer DW, Berger JN, et al. (2016) Suppression of antitumor immune responses by human papillomavirus through epigenetic downregulation of CXCL14. MBio 7(3): е00270-16.
-
Sotlar K, Koveker G, Aepinus C, Selinka HC, Kandolf R, et al. (2001) Human papillomavirus type 16-associated primary squamous cell carcinoma of the rectum. Gastroenterology 120(4): 988-994.
-
Fahey LM, Raff AB, Da Silva DM, Kast WM (2009) A major role for the minor capsid protein of human papillomavirus type 16 in immune escape. J Immunol 183(10): 6151-6156.
-
Molina AA, Valencia JFH, Lamoyi E, Paredes AC, Lizano M (2013) Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses 5(11): 2624-2642.
-
Orange JS (2013) Natural killer cell deficiency. J Allergy Clin Immunol 132(3): 515-526.
-
Handisurya A, Day PM, Thompson CD, Bonelli M, Lowy DR, et al. (2014) Strain-specific properties and T cells regulate the susceptibility to papilloma induction by Mus musculus papillomavirus 1. PLOS Pathogens 10(8): e1004314.
-
Uberoi A, Yoshida S, Frazer IH, Pitot HC, Lambert PF (2016) Role of ultraviolet radiation in papillomavirus- induced disease. PLOS Pathogens 12(5): e1005664.
-
Jackson SE, Mason GM, Wills MR (2011) Human cytomegalovirus immunity and immune evasion. Virus Research 157(2): 151-160.
-
DiMaio D, Petti LM (2013) The E5 proteins. Virology 445(1-2): 99-114.
-
Campo MS, Graham SV, Cortese MS, Ashrafi GH, Araibi EH, et al. (2010) HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 407(1): 137-142.
-
Miura S, Kawana K, Schust DJ, Fujii T, Yokoyama T, et al. (2010) CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV. J Virol 84(22): 11614- 11623.
-
Ashrafi GH, Haghshenas M, Marchetti B, Campo MS (2006) E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int J Cancer 119(9): 2105-2112.
-
Seitz H, Schmitt M, Bohmer G, Schneider AK, Muller M (2012) Natural variants in the major neutralizing epitope of human papillomavirus minor capsid protein L2. International Journal of Cancer 132(3): Е139-148.
-
Strickler HD, Burk RD, Fazzari M, Anastos K, Minkoff H, et al. (2005) Natural history and possible reactivation of human papillomavirus in human immunodeficiency virus-positive women. J Natl Cancer Inst 97(8): 577-586.
-
Wang CJ, Sparano J, Palefsky JM (2017) Human immunodeficiency virus/AIDS, human papillomavirus, and anal cancer. Surgical Oncology Clinics of North America 26(1): 17-31.
-
Bonnet F, Chene G (2008) Evolving epidemiology of malignancies in HIV. Current Opinion in Oncology 20(5): 534-540.
-
Shiels MS, Cole SR, Kirk GD, Poole C (2009) A meta- analysis of the incidence of non-AIDS cancers in HIV- infected individuals. Journal of Acquired Immune Deficiency Syndromes 52(5): 611-622.
-
Williamson AL (2015) The interaction between human immunodeficiency virus and human papillomaviruses in heterosexuals in Africa. Journal of Clinical Medicine 4(4): 579-592.
-
Scott M, Nakagawa M, Moscicki AB (2001) Cell-mediated immune response to human papillomavirus infection. Clinical and Diagnostic Laboratory Immunology 8(2): 209-220.
-
Alkhatib G (2009) The biology of CCR5 and CXCR4. Current Opinion in HIV AIDS 4(2): 96-103.
-
Burger JA, Kipps TJ (2006) CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood 107(5): 1761-1767.
-
Ansari AW, Heiken H, Moenkemeyer M, Schmidt RE (2007) Dichotomous effects of C-C chemokines in HIV-1 pathogenesis. Immunol Lett 110(1): 1-5.
-
Rice AP (2017) The HIV-1 Tat protein: mechanism of action and target for HIV-1 cure strategies. Curr Pharm Des 23(28) 4098-4102.
-
Palefsky J (2006) Biology of HPV in HIV infection. Adv Dent Res 19(1): 99-105.
-
Das AT, Harwig A, Berkhout B (2011) The HIV-1 Tat protein has a versatile role in activating viral transcription. J Virol 85(18): 9506-9516.
-
Syrjanen S (2011) Human papillomavirus infection and its association with HIV. Adv Dent Res 23(1): 84-89.
-
Barillari G, Palladino C, Bacigalupo I, Leone P, Falchi M, et al. (2016) Entrance of the Tat protein of HIV-1 into human uterine cervical carcinoma cells causes upregulation of HPV-E6 expression and a decrease in p53 protein levels. Oncol Lett 12(4): 2389-2394.
-
Nyagol J, Leucci E, Onnis A, De Falco G, Tigli C, et al. (2006) The effects of HIV-1 Tat protein on cell cycle during cervical carcinogenesis. Cancer Biol Ther 5(6): 684-690.
-
Nakagawa M, Stites DP, Palefsky JM, Kneass Z, Moscicki AB (1999) CD4-positive and CD8-positive cytotoxic T lymphocytes contribute to human papillomavirus type 16 E6 and E7 responses. Clin Vaccine Immunology 6(4) 494-498.
-
Welters MJ, Kenter GG, Piersma SJ, Vloon AP, Lowik MJ, et al. (2008) Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res 14(1): 178-187.
-
Maskey N, Thapa N, Maharjan M, Shrestha G, Maharjan N, et al. (2019) Infiltrating CD4 and CD8 lymphocytes in HPV infected uterine cervical milieu. Cancer Management and Research 11: 7647-7655.
-
Yaghoobi M, Le Gouvello S, Aloulou N, Duprez DC, Walker F, et al. (2011) FoxP3 overexpression and CD1a+ and CD3+ depletion in anal tissue as possible mechanisms for increased risk of human papillomavirus-related anal carcinoma in HIV infection. Colorectal Dis 13(7): 768- 773.
-
Meier A, Bagchi A, Sidhu HK, Alter G, Suscovich TJ, Kavanagh DG (2008) Upregulation of PD-L1 on monocytes and dendritic cells by HIV-1 derived TLR ligands. AIDS 22(5): 655-658.
-
Sobhani I, Walker F, Aparicio T, Abramowitz L, Henin D, et al. (2002) Effect of anal epidermoid cancer-related viruses on the dendritic (Langerhans) cells of the human anal mucosa. Clin Cancer Res 8(9): 2862-2869.
-
Allouch S, Malki A, Allouch A, Gupta I, Vranic S, et al. (2020) High-risk HPV oncoproteins and PD-1/PD-L1 interplay in human cervical cancer: recent evidence and future directions. Front Oncol 10: 914.
-
Keir ME, Butte MJ, Freeman GJ, Sharpe AH (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26: 677-704.
-
Matloubian M, Concepcion RJ, Ahmed R (1994) CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68: (12) 8056-8063.
-
Deeks SG, Tracy R, Douek DC (2013) Systemic effects of inflammation on health during chronic HIV infection. Immunity 39(4): 633-645.
-
Gulzar N, Copeland KF (2004) CD8+ T-cells: function and response to HIV infection. Current HIV Research 2(1): 23-37.
-
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, et al. (2006) PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443(7109): 350-354.
-
Cockerham LR, Jain V, Sinclair E, Glidden DV, Hartogenesis W, et al. (2014) Programmed death-1 expression on CD4+ and CD8+ T cells in treated and untreated HIV disease. AIDS 28(12): 1749-1758.
-
Porichis F, Kaufmann DE (2012) Role of PD-1 in HIV pathogenesis and as target for therapy. Current HIV/ AIDS Reports 9(1): 81-90.
-
Grabmeier PK, Steinberger P, Rieger A, Leitner J, Kohrgruber N (2011) Identification of PD-1 as a unique marker for failing immune reconstitution in HIV-1- infected patients on treatment. Journal of Acquired Immune Deficiency Syndromes 56(2): 118-124.
-
Papasavvas E, Surrey LF, Glencross DK, Azzoni L, Joseph J, et al. (2016) High-risk oncogenic HPV genotype infection associates with increased immune activation and T cell exhaustion in ART-suppressed HIV-1-infected women. Oncoimmunology 5(5): е1128612.
- Are the Vaccines the Only Solution to Prevent the COVID-19 Pandemic? Part Two
- Clinical Characteristics of Women in this New Global Immunodeficiency
- Cell Dynamics in HIV Pathogenesis: Insights and Implications
- Determination of the CDR (CDR1, CDR2) « Complementary- Determining Region Invertebrate Primitive Antibody from Sea Star »
- Prioritizing Care for High-Risk COVID-19 Patients in the EU: 10 Civic Recommendations to the Institutions
- Comprehensive Insights into ModRNA Vaccines: Persistent PP-Spike Recombinant Protein, Hyperimmune/Inflammatory Reactions, Thrombotic Vasculopathy, Chronic Organ Complications and Excess Deaths