Chronic Hypersensitivity Pneumonia. What the Clinical Radiologist Participating in a Multidisciplinary Meet Needs to Know
Chronic hypersensitivity pneumonia (CHP) is a diffuse interstitial lung disease, brought about by an inflammatory response to various known and unknown antigens. It affects the small airways, its surrounding parenchyma and interstitium. It can manifest either as a fibrosing or non-fibrosing diffuse lung disease. The fibrotic pattern is irreversible, hence if not recognized early, it embarks on a downhill course. The pattern and anatomical distribution of fibrosis shows some differences from other fibrosing disorders. Features resembling sarcoidosis, fibrosing NSIP or UIP may be seen towards the end stage, making distinction difficult. Clinical evaluation and perusal of previous records helps suggest the diagnosis and assess its progress. In this review article, an attempt will be made to understand the disease in order to enable the radiologist add value in a multidisciplinary discussion (MDD).
Introduction
Chronic hypersensitivity pneumonia is a diffuse fibrosing lung disease initiated by exposure to a variety of stimulants, both organic and inorganic, some being finer than 5 μm [1]
in size. The body’s immune mediated response to the inhaled antigen [2] initially results in acute inflammatory changes which if not managed promptly, progresses to diffuse fibrosis. This disease has serious implications on the health and longevity of the patient. Unfortunately, despite being common, it is either misdiagnosed or underdiagnosed. It is therefore, important to bring it upfront in the ILD conundrum so that appropriate management protocols can be instituted, at the earliest. The radiologist can play an important role in a MDD by suggesting its possibility and commenting on its progression following comparison with prior studies.
Clinical Aspects
The recently initiated Indian registry for ILD has shown hypersensitivity pneumonia (HP) as the second most common ILD in India (24% of total ILDs) [3]. It is also the second commonest ILD in Brazil [4] and the third commonest ILD in Denmark [5]. The described incidence ranges from 0.3 to 0.9 per 100,000 population [5, 6] and increases to 18-50% [7] in new onset ILDs, following an MDD. The prevalence of HP has been increasing on account of prolonged and continuous exposure to myriads of known and unknown antigenic stimuli. Unfortunately, despite thorough investigation [8], in 50 to 60 % cases, the cause remains unidentifiable [9].
Compared to the general population, the incidence is higher amongst bird feeders [10]. The stimulus in these patients could be a mix of antigens rather than a single offending agent, as not everyone exposed to birds, develops CHP. There may be interplay of multiple factors, including genetic predisposition, leading to greater effect of the exposure on them, as compared to others [11]. Thus, obtaining family history is important, especially if the patient is young [12].
The mean age of presentation varies. . It was 55 years +/- 11 years in a recently published Indian study [3]. Amongst patients under 18 years, males were affected more often, while above 18 years, the incidence was more in females. The age of onset is lower in some communities who are pigeon feeders, racers and breeders. The radiologist must insist on a detailed history with respect to occupation, hobbies and environment of the patient. It will increase the level of confidence while offering this diagnosis.
Pathogenesis
The pathogenesis of the disease suggests both type III (immune complex–mediated) and type IV (delayed) hypersensitivity reactions to an inciting agent [13]. The inciting antigens can be proteins derived from the living world, polysaccharides or low molecular weight non-protein chemicals [14].
Smoking and Hypersensitivity
Smoking plays a paradoxical role in the course of hypersensitivity pneumonia (HP). HP develops less frequently in smokers as compared to non-smokers. This is because, initially, smoking reduces the risk and severity of CHP [15] by reducing the body’s inflammatory response on the smoker’s lungs. In the long term, however, this very lack of protective effect of the body’s defense mechanism, leads to a downhill clinical course [15].
Bronchioalveolar Lavage (BAL)
Bronchioalveolar lavage done immediately following an exposure or during acute exacerbation may show raised neutrophil count [16]. The initial neutrophil increase is followed by lymphocytosis which can be over 30% [17]. The lymphocyte count can even increase to up to 60-80% (N: 15% or less) The “Cluster of Differentiation” (CD), CD4 to CD8 ratio is low. The CD4+/CD8+ lymphocyte ratio is less than 1, because of the increase in numbers of CD8+ cells. In these patients, Natural Killer T (NKT) cells are also elevated in BAL fluid. However, neither lymphocytosis nor a low CD4: CD8 ratio is sufficiently specific. Tissue diagnosis may be needed to establish the diagnosis [18].
Histopathological Features
The disease is broncho-centric centric (Figure 1). As it advances, the twin processes of inflammation and repair extends beyond the airways to involve the adjoining parenchyma and interstitium [19]. This progression causes airway constriction, airspace attenuation, architectural distortion, and occlusion of blood vessels in the center as well as periphery of the secondary pulmonary lobule.
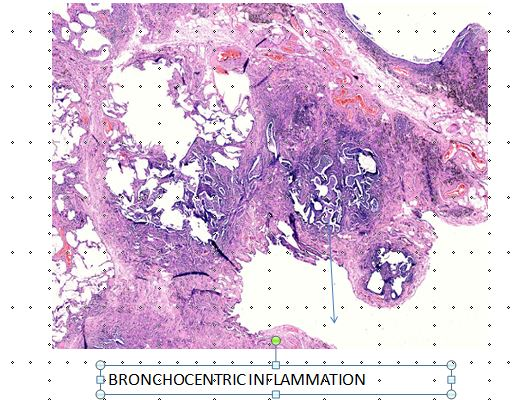
Amongst the specimen obtained by various methods of biopsies, transbronchial biopsy specimen may not be very helpful. They may be nonspecific in up to 48% cases [20]. However, the presence of diffuse lymphocyte infiltration may be a clue towards HP [20]. Biopsy specimen from a single site may also be inadequate. A granuloma may not be present in one biopsy specimen [21]. Biopsies from multiple sites may be needed to ascertain the diagnosis [22]. Surgical lung biopsies are, therefore, more sensitive, as a larger number of samples from both affected and non-affected areas, can be obtained. The classical features of chronic HP in lung biopsy, include presence of ill-formed granuloma, bridging or non- bridging as well as peribronchial fibrosis, peribronchiolar lymphocyte predominance and peribronchiolar organization pneumonia. The radiologist should provide inputs regarding the appropriate sites for obtaining biopsies.
Granulomas
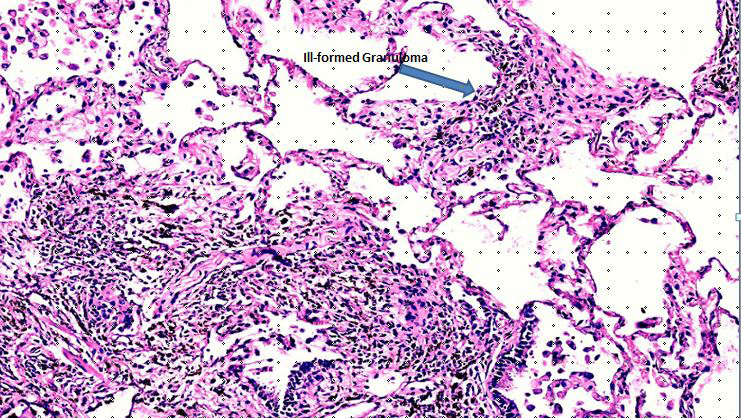
Granulomas of HP are ill formed, non-caseating, aggregates of lymphocytes, plasma cells, macrophages and multinucleated giant cells (Figure 2). The multinucleated giant cells may show presence of nonspecific cytoplasmic inclusions such as Schaumann bodies, asteroid bodies, cholesterol clefts, in addition to birefringent calcium salts [20]. The granulomas may be imperceptible from the surrounding interstitial infiltrates. The peribronchial location of granulomas is typical for HP [21]. Rarely a granuloma may be endobronchial in location [13]. Infact, in transbronchial biopsy specimen, granulomas may even be found within bronchiolar walls [5].
Fibrosis
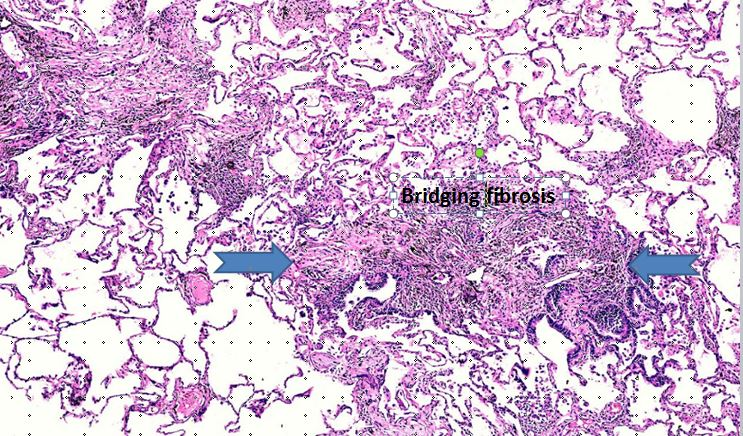
Fibrosis may be non-bridging or bridging (Figure 3), that is, it can connect neighboring centrilobular areas [23] or stretch between centrilobular and perilobular regions [24]. As the disease progresses, the fibrosis may become indistinguishable from fibrotic NSIP or UIP pattern [25].
Bronchiolitis
The accompanying inflammation is broncho-centric and is indicative of active small airways component of the disease process. The composite findings of interstitial fibrosis, ill formed granulomas and cellular bronchiolitis may be seen in up to 80% cases of CHP [26]. There may also be findings of organizing pneumonia and lymphocytosis. Fibrin deposition, eosinophils and neutrophils and necrotizing vasculitis are usually suggestive of co-existing acute phase of hypersensitivity pneumonia [27].
The histological findings of HP may mimic several diseases, including early sarcoidosis, drug reaction, LIP. Often, the pathologist may only be able to provide a descriptive report [13]. The final diagnosis may be arrived only through MDD.
Clinical Course
CHP is characterized by progressive increase in respiratory effort, with deterioration of lung functions, culminating into oxygen dependency and respiratory failure. The disease can appear insidiously [20]. The progress may be from acute to subacute to the chronic stage, known as chronic hypersensitivity pneumonia. Hence the descriptors, acute, subacute or chronic stages of the disease process, are used. The acute stage could begin within 4 to 24 hours of exposure and manifest as viral pneumonia. The subacute phase occurs over a period of weeks or months with the symptoms appearing intermittently. Persistence of exposure and therefore symptoms beyond six month, leads to the chronic stage. The patient may be intermittently sick or suffer from prolonged, severe dyspnea and malaise.
The categorization into three separate groups suggests clear division of the stages, particularly the subacute stage [20] which is really not the case. A recent clinical classification system, which reduces the three stages to two, namely “acute” often reversible HP lasting less than 6 months and “chronic” irreversible HP lasting beyond 6 months, has been suggested [11]. The ATS guideline committee [22] has also recently categorized HP on the basis of CT findings as either fibrotic (i.e., mixed fibrotic plus inflammatory or purely fibrotic) or non-fibrotic (i.e., purely inflammatory). When the patient has mixed features, the categorization is determined by the dominant findings in the HRCT.
Pulmonary Function Tests (PFT)
The three main components considered in CHP are, (a) total lung capacity (TLC), which is reduced and residual volume (RV), which is increased), (b) evaluation of dynamic lung functions by spirometry and flow volume loop (c) and diffusion capacity of the lung for carbon monoxide (DLCO), which is also reduced [28]. In CHP, we can expect to see reduced FEV1/ FVC ratio. The value of PFT is in evaluating the status of lung function at a given time point and in follow-up [15]. It has been found that a decline of forced vital capacity (FVC) by 10% or more and DLCO by 15 % or more, within the first year, is associated with increased mortality [29].
Radiographic Findings
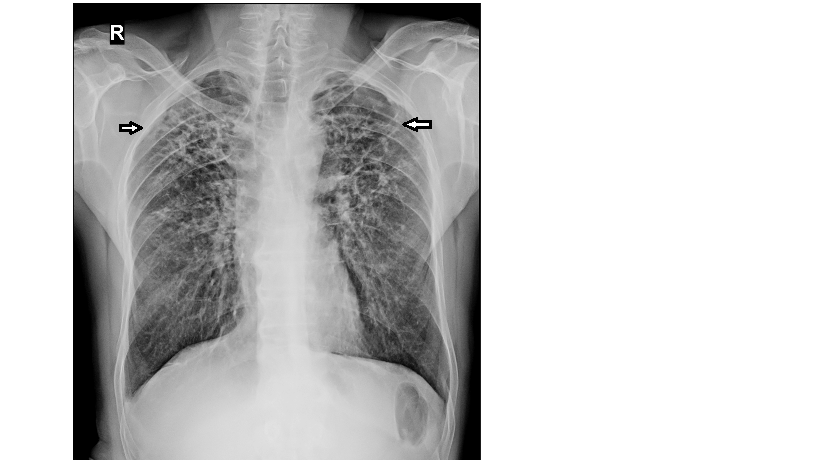
Plain x-ray chest findings in CHP are non- specific but useful (Figure 4). X-rays may show evidence of upper lobe fibrosis causing ipsilateral tracheal retraction and parenchymal distortion. Serial x-rays must be obtained for comparison, to evaluate for any shrinkage of the lungs. The outlines of the diaphragms, pleura and pulmonary vessels become indistinct with ensuing fibrosis.
Ultrasound
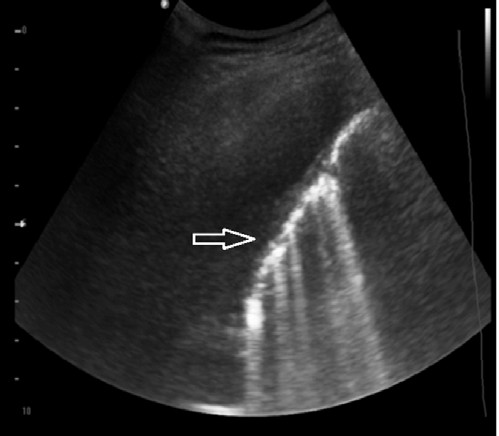
Ultrasound may show increased number of “B” line due to focal areas of parenchymal consolidation or fibrotic lesions. Distinct B lines suggest focal or scattered fibrosis (Figure 5). Coalescent B lines suggests consolidation or edema. Diaphragmatic excursion and height are also affected by fibrosis and volume loss [30, 31]. Reduced diaphragmmatic excursion, spells a bad prognosis. High position, suggests fibrosis.
High resolution CT (HRCT) of the Chest
The present day multidetector scanners (MDCT) obtains data from a volume of lung tissue. An HRCT image (thinner sections using an edge enhancement) can easily be sliced off from this dataset for analysis of the pulmonary interstitium.
Technical Aspects
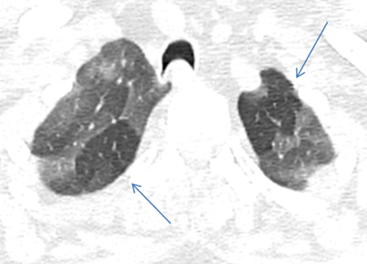
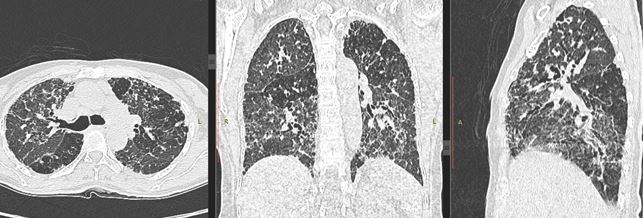
A non-contrast CT scan (NCCT) should be obtained in inspiratory and expiratory phases. A prone position scan may occasionally be needed [21]. The expiratory scans enable detection of air trapping by highlighting well demarcated low attenuating areas with sparse blood vessels, provided the fibrosis is not extensive (Figure 6). The affected lobules also appear as “simplified” areas within the distorted lung parenchyma due to hypoxic vascular constrictive effect (Figure 7). Minimal intensity projections (MinIP) images with an aperture width of 2-3 cms, are helpful in identifying areas of air trapping (Figure 8).
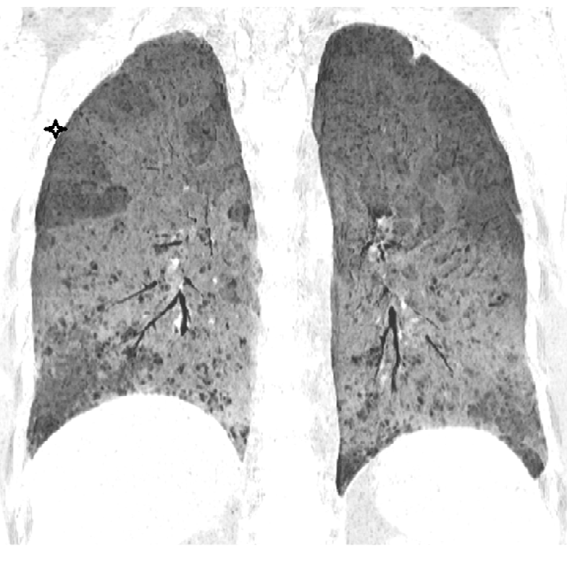
It is advantageous to evaluate scans in multiple planes for a clearer idea of the distribution of the disease (Figure 9).
Low dose CT (LDCT) with edge enhancing kernels can be performed because patients might undergo multiple CTs during follow up. The images are of adequate quality for evaluation the pulmonary interstitium. A kernel using smoother settings can be also applied for examination of the mediastinal structures and bones. IV contrast administration is usually not needed, unless there is a suspicion of pulmonary embolism, PAH or any abnormal shadow is present in NCCT. In case IV contrast administration is planned, a non-contrast scan should be obtained first. This is because contrast leads to a ground glass like enhancement of the lung tissue, thereby suggesting acute exacerbation or presence of non-fibrotic disease process.
HRCT Features of CHP
These include several of the under mentioned findings: Fibrosis: Fibrosis develops after several months of continuous or repeated exposure to antigens. Usually, it takes about 6 months to develop [14].
Thereafter, the radiological findings may show stability, slow progress or rapid worsening, irrespective of duration of exposure or treatment. The fibrosis is thick, dense, coarse (Figure 10), predominantly peribronchial with peripheral reticulations or perilobular (Figure 11) leading to asymmetric parenchymal distortion and traction on the trachea, bronchial and vessel walls.
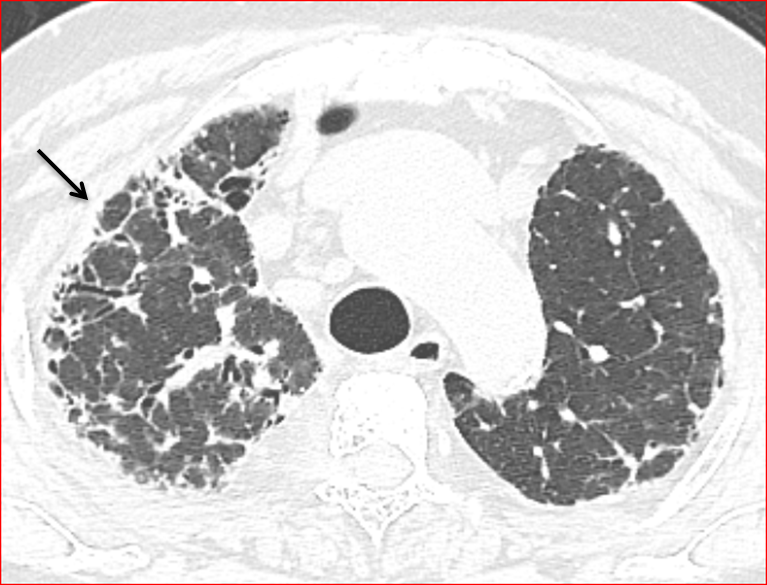
The axial spread is around the peribronchovascular structures, extending outwards from the hila towards the lung periphery, often like a spoke wheel (Figure 12). It is seen predominantly in the upper and mid zones [32], but can involve any or all zones the lower zones are comparatively less involved. However, some articles have also reported a mid [32] as well as lower zone predominance, to an extent of up to 30-50% in the lower lobes [9]. The upper zone involvement has even been reported to be not as commonly affected as is believed, the incidence being < 10% [22]. Therefore, though more likely to be found in the upper lobes, the distribution of fibrosis might, not be a clincher in the diagnosis of CHP.
The fibrosis is bilateral, asymmetrical with relative sparing of the extreme apices and bases [32]. However, no area is immune to involvement by the fibrosis (Figure 13).
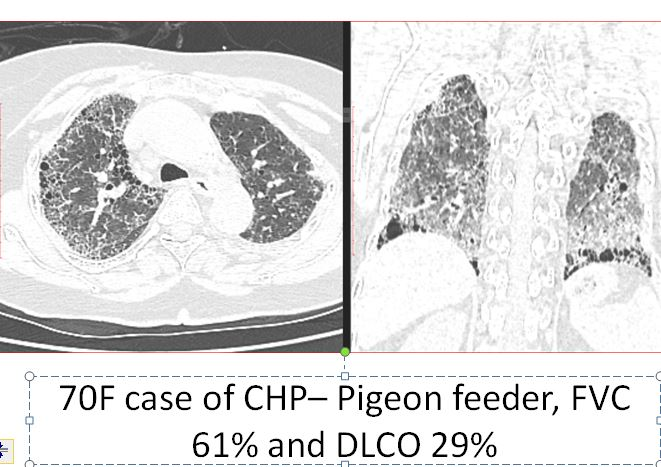
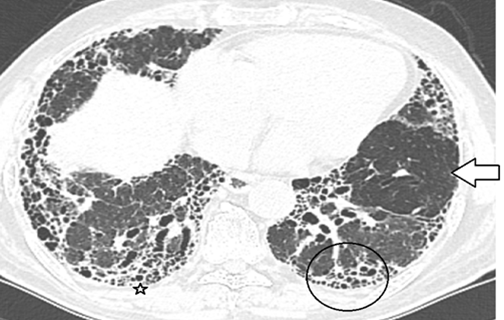
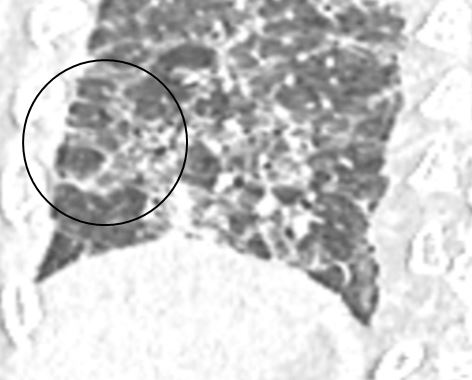
Whether central or peripheral, fibrosis results in traction bronchiectasis, bronchiolectasis and honeycomb cystic changes (Figure 14 a, b) .It may give a constrictive bronchiolitis like appearance [33] by involving the small peripheral non-cartilaginous lobular bronchi, located distal to the terminal bronchioles. These airways normally, not visible may, become appearing as cystic or tubular branching structures. There may be honeycombing, in 16-69 % of cases [34], which can be seen in any lobe, not necessarily in the posterobasal region as it happens in IPF.
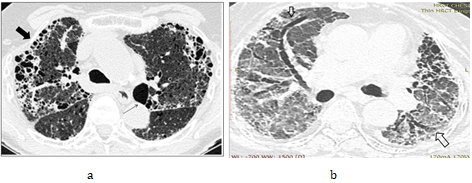
Figure 14 a, b: showing honeycomb cysts (thick arrow), bulla (thin arrow) and traction bronchiectasis (outlined arrow).
Infact, when honeycombing involves the lower lobes, distinction from IPF UIP becomes difficult even for an experienced thoracic radiologist. The peripheral fibrosis can cause pleural retraction with fatty replacement in the extrapleural spaces. The radiologist must also be aware of the fact that occasionally, chronic HP may be non- honeycomb type in which case it becomes difficult to distinguish from non-honeycomb UIP IPF. In such cases, the MDD holds utmost importance.
The differentiation of CHP from other causes of fibrosis is only possible in approximately 50 % of patients only. It is helpful if there is presence of mosaic pattern, centrilobular nodules and lack of lower zone predominance of the fibrosis [34]. These features may be present in prior imaging studies and not be seen at a later stage. Hence examination of prior studies is important.
Mosaic pattern: Chronic HSP, being predominantly an airway obstructive disorder with secondary peribronchial and parenchymal fibrosis, causes a ball valve type of occlusion, resulting in varying degrees of air trapping. Since fibrosis is often seen in the upper lobes, lobular or sub-segmental air trapping in the upper lobes, favors a diagnosis of chronic HP .Air trapping in the lower lobes can be difficult to observe, if the patient is unable to hold breath during scanning.
The increased lucency is however not simply due to a ball valve obstruction of the bronchioles. Air continues to enter the alveolar spaces through intraalveolar pores of Kohn, the bronchioloalveolar canals of Lambert and interbronchiolar channels of Lambert [35], but is unable to exit ,thereby increasing the volume as well as lucency of the affected lobe.
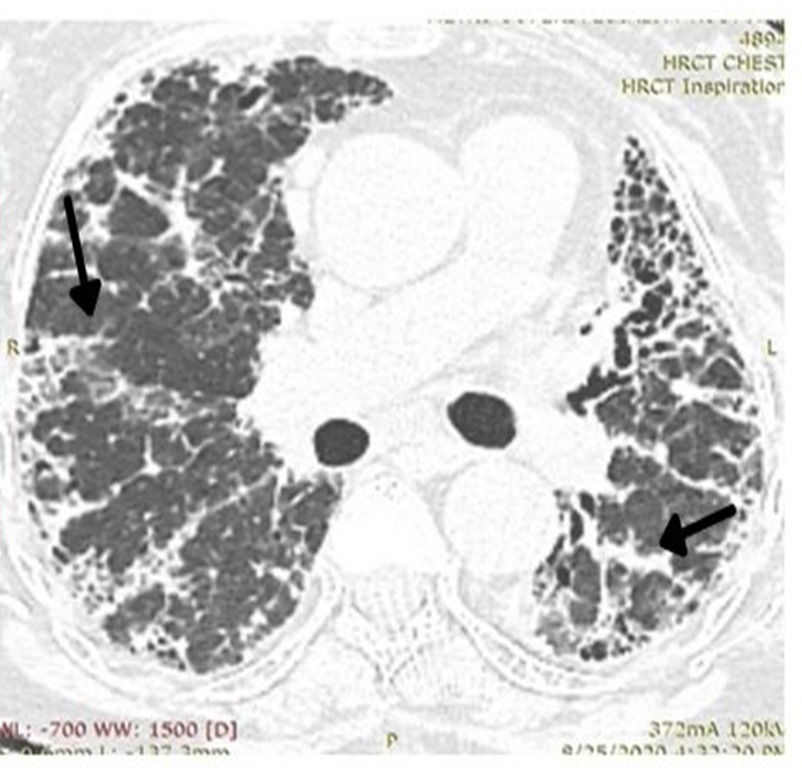
Another factor contributing to the lucency, is that vascular constriction occurs due to hypoxia, and results in redistribution of blood to the aerated regions [36]. This phenomenon of redistribution is a mechanism to maintain gas exchange. The presence of “simplified areas” of lung, that is, areas showing paucity of vascular marking, is a useful marker of air trapping (Figure 15). These areas, distended with trapped air, compress and cause ground glass haziness and crowding of the vessels in the adjacent pulmonary lobules, thereby increasing their conspicuity.
As the name suggests, a mosaic pattern on HRCT is a formed by a cluster of pulmonary parenchymal tissue having different densities. There are areas of normal lung density ground glass opacity and lucency- called the triple density sign. The observation of mosaic attenuation depends on the extent of fibrosis. In fact, the greater the fibrosis, the lesser the visibility of the mosaic pattern [37].
Inability to exhale adequately, adds to the difficulty in demonstrating mosaic pattern in the expiratory phase. In order to be able to detect areas of air trapping, which should be at least in more than 5 lobules [22], a proper expiratory scan is needs, which may not be feasible in a breathless patient. So, it is important to be able to recognize air trapping in whichever phase of breath-hold, the image is obtained, by looking out for “simplified areas of lung parenchyma” [38]. (A “simplified area, differs from “normally aerated lung parenchyma” as it shows comparatively reduced or attenuated vascular markings). It should also be borne in mind that mosaic attenuation can result from coexisting bronchial pathology as well as by the presence of inflammatory fluid in some areas, while sparing others [39].
Mosaic attenuation may also be seen in cases of IPF, UIP
due to other causes and in Sarcoidosis [22]. In these diseases, mosaicism may be less compared to CHP [37]. A sign of air trapping, specific for chronic fibrotic HP [40] is called a “head cheese sign/ triple density sign” (Figure 16). This is produced by a combination of densities, made increased lucency due to air trapping, ground glass opacity to due inflammation and normal lung density.
Of note, is the importance of remembering that slight mosaicism may also be seen in up to 20% of normal individuals including smokers.
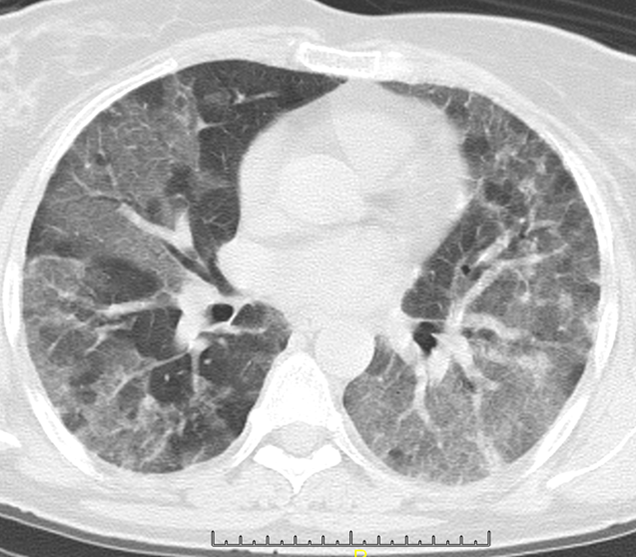
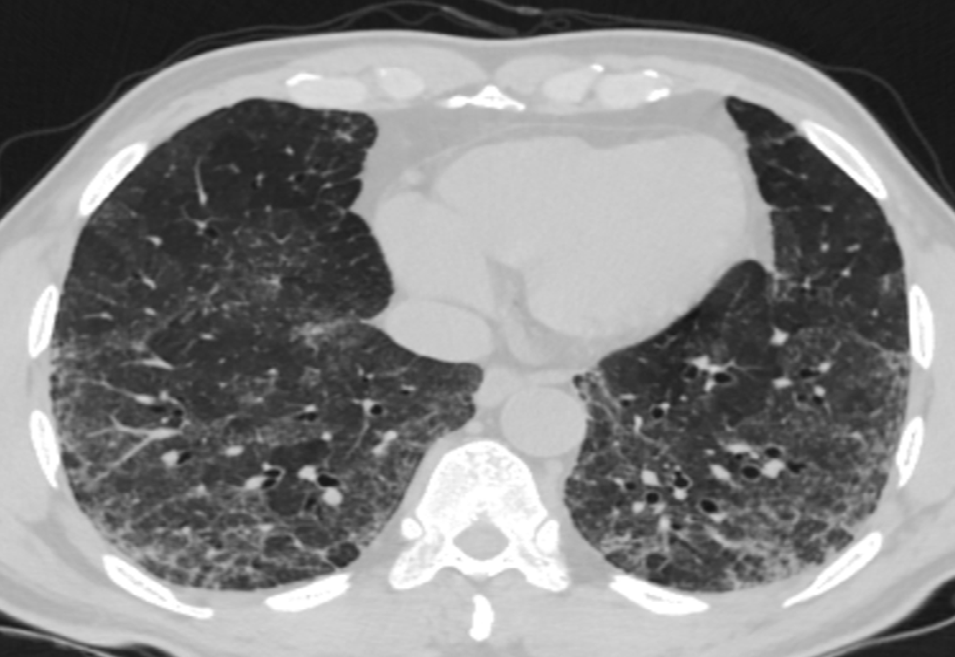
Ground Glass (GG) Opacities: Ground glass opacity could either be post inflammatory or sequelae to irreversible intralobular fibrosis. Post inflammatory GG opacity could be due to both lymphocytic infiltration and minor degree of organization (Figure 17) [34].
When fibrosis is the cause of the ground glass opacity, it’s intralobular location results in traction bronchiolectasis. This bronchiolectasis, can be observed in the periphery of the lung fields, enabling distinction of fibrotic from inflammatory pathology. Ground glass opacity is however, more commonly a feature of a coexisting acute process [41].
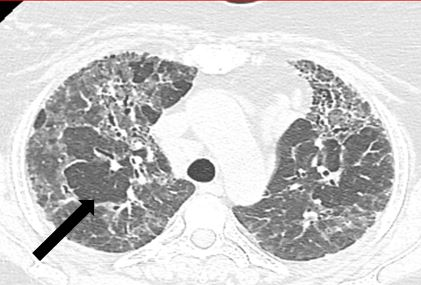
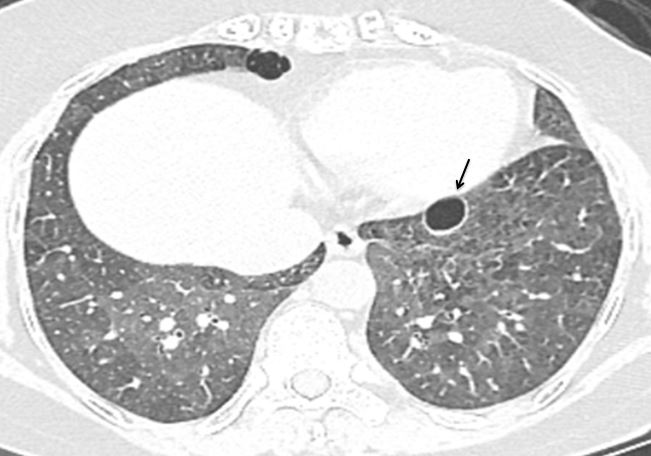
Cysts and Emphysematous Changes: Thin walled cysts are an occasional finding in chronic HP. These are perivascular cysts caused by obstructed bronchi (Figure 18). They are sparse and randomly distributed.
Emphysematous changes may also be seen in some patients. It is thought that airway obstruction which is a reason for development of perivascular cysts [42] also result in formation of emphysematous bulla [43]. Therefore, if a non-smoker shows features of emphysema, it should elicit concern for chronic HP. Features of emphysema rather than fibrosis [44] are commonly seen in nonsmokers with farmer’s lung [45]. Emphysema may range from mild to severe, may be centrilobular or paraseptal [46] and may even present as a focal bulla. Though commoner in the upper lobe, any lobe could be affected. Combined pulmonary fibrosis with emphysema (CPFE), like features, may also be seen in cases of CHP with a UIP (honeycomb) phenotype.
Pulmonary Hypertension
Pulmonary hypertension is a known adverse complication of chronic HP and is related to the severity of fibrosis [47]. There are various mechanisms which can cause PAH. These include pulmonary vasoconstriction, pulmonary vascular remodelling, small vessel destruction, and fibrosis [48]. The proximity of fibrosis to the peribronchovascular structures may also be a contributory factor [49]. Inhaled antigens acting as vasoconstrictors, also be a as a causative factor [50]. The pulmonary hypertension is pre-capillary type and comes under group 3 of the International Etiological Classification of Pulmonary Hypertension [51]. Symptomatic cases of CHP with extensive fibrosis may result in pulmonary hypertension in up to 44% cases [50].
CT can give useful information regarding PAH. Raised pulmonary artery pressure is manifested by dilatation of the MPA (more than 3 cms or greater than ascending aorta) followed by dilatation of the right ventricle, straightening or bowing of interventricular septum towards the LV, enlargement of the RA and development of tricuspid regurgitation. Cardiac decompensation may ensue, as manifested by development of cardiomegaly and pleural effusion. The implication of PAH in a case of CHP is towards prognostication and management issues like consideration of lung transplant.
Lymphadenopathy
Lymph node enlargement may be seen infrequently in CHP [52]. Both mediastinal and hilar nodes [53] may be enlarged. However, bilateral hilar lymphadenopathy alone is not seen with chronic HP [54].On histopathology, the nodes may be reactive or show non-necrotizing granulomas [55]. Granulomas have been reported in the mediastinal nodes in patients of farmer’s lungs [32]. The nodal enlargement is mild, not clumped or mass- like with no pressure effect on the adjoining structures. The nodes do not show calcification, enhancement or necrosis. The presence of enlarged mediastinal or hilar nodes with any associated feature should prompt exclusion of diseases such as TB, Sarcoidosis and malignancies.
Comorbidities Associated with Chronic HP
The radiologist should be on the lookout for atherosclerotic changes, coronary artery calcification, esophageal dilatation, hiatus hernia. The density of the thyroid gland should be evaluated on NCCT. If density is less than or equal to adjoining muscles, it should be correlated with thyroid function tests. Patients having hypothyroidism and having a high BMI fatigue easily and may develop exertional dyspnea [56].
The high BMI [5]. can be suspected by examining the body fat included in the NCCT study. The radiologist should also comment on muscle mass. A sacropenic patient may not do well. Lung cancers can develop near an area of honeycombing, especially in smokers [57]. These lesions could be mistaken for focal organization, if solid or mimic acute exacerbation, if they show a ground glass pattern. Patients may have had Covid 19 infection. The interstitial lung abnormality in long haulers Covid patients may merge with the underlying fibrosis of CHP, making distinction difficult.
Acute exacerbations (AIP) of CHP are commoner than superadded infections [58] including viral infections. It should also be remembered that a patient of CHP, on steroids, or having diabetes could develop tuberculosis, fungal or some other atypical infection. Since most pulmonologists focus mainly on lungs, it makes the role of a physician radiologist very, important in patient care.
Prognostic Factors in Imaging
The varied clinical and radiological features of chronic HP, makes it imperative to be certain of the diagnosis. A confident diagnosis is important as further management and prognosis is based on the level of certainty. An algorithm has been proposed to define the confidence / certainty level of the diagnosis of CHP, meaning that if the level of confidence in the clinico-radiological diagnosis is very high, then a confirmatory biopsy may be avoidable. The level of confidence is divided into 4 groups, namely group 1- confident, group 2-probable, group 3-possible and group 4- unlikely diagnosis [14].
Although this algorithm is helpful, it is subjective, hence definite diagnosis still depends upon histopathological markers. Once a diagnosis of CHP is firmly established, the clinicoradiological prognostic factors should be looked into.
The prognosis is generally poor with a 5-year survival of 71–82% [59, 6]. Fibrosis and traction bronchiectasis is associated with a poor prognosis [39, 60]. An attempt has been made to prognosticate based on 3 patterns of CHP [61]. These patterns are honeycomb cysts, fibrosis without honeycombing and non-fibrotic patterns. Among these phenotypes, the honeycomb type has the worst prognosis, whiles the non-fibrotic, has the best prognosis. Since the presence of honeycombing indicate a poor prognosis, when seen, further invasive diagnostic procedures are of limited value. .
The presence of air trapping with a mosaic pattern suggests a better prognosis [9]. Presences of coexistent autoimmune features have also been shown to be an independent predictor of poor prognosis [62]. 15% of patients with chronic HP display coexistent autoimmune disease or autoimmune features. These patients are considered to have a bad prognosis [63]. Emphysematous changes along with HP do not seem to be a significant prognostic factor. CPFE has also been shown to be an independent predictor of mortality.
Pulmonary hypertension is an important prognostic factor. The presence of increased neutrophils in BAL has shown correlation with the development of fibrosis and, therefore might be a predictor of poor outcome [64]. Smokers are initially less likely to develop HP, as they have a lower level of specific antibodies to the causative agent. However, once HP sets in, the course is downhill with a poor clinical outcome [65]. The presence of coexisting cardiovascular disease has a significant impact on survival in CHP. Hence it is necessary to keep this factor in mind while prognosticating [66]. Older age and a poor baseline pulmonary function is also an indicator of poor prognosis [62].
The overall survival rate for both males and females patients of chronic HP, is lower as compared to matched general population [67]. The mean survival rate for chronic HP is between 7 to 9 years which is better than IPF. The deaths are mainly due to respiratory or cardiac cause and are increased in the initial 2 years following the diagnosis [67].
Differential Diagnosis of CHP
Amongst many possible causes of diffuse fibrosing lung disease, there are some which predominantly affect the upper lobes. These include sarcoidosis, tuberculosis, histoplasmosis, chronic allergic bronchopulmonary fibrosis, silicosis, ankylosing spondylosis, drug related toxicity. Others, such as NSIP fibrosis, UIP IPF and connective tissue disease related ILD, affect predominantly the lower lobes. The radiologist would do well not to overlook the many other mimics of CHP while participating in the MDD.
Conclusion
Chronic Hypersensitivity pneumonitis (HP) is a diffuse fibrosing lung disease resulting from repeated exposure to a variety of known and unknown particulate matters. It could be non-fibrosing or fibrosing with overlaps of acute exacerbation.
Pathologically, CHP is characterized by presence of ill- formed granuloma, and peribronchial fibrosis. On HRCT scan, coarse reticular opacities, volume loss, and traction bronchiectasis with mosaic pattern may be observed. Importantly, CHP may mimic or progress into other fibrotic forms, including NSIP and UIP, making etiological differentiation difficult in the late stage. Thus, CHP requires a high index of suspicion and should be considered in any patient presenting with clinical evidence of interstitial lung disease. A definitive diagnosis requires clinical, radiologic, laboratory, and pathologic correlation. Early diagnosis and avoidance of further exposure are keys in management of the disease. Corticosteroids are generally used, although their long-term efficacy has not been proven in prospective clinical trials. Immunosuppressant, antifibrotics and lung transplantation may be considered in cases of progressive illness [1].
The role of the radiologist is to suspect this disease in the early stages, differentiate it from other fibrosing lung diseases, look out for acute exacerbations, other comorbidities and determine course of the disease if serial studies are available. The radiologist can provide valuable input to both the pathologist and pulmonologist, in a MDD, thus helping in better patient management.
References
-
Selman M, Roldan IB (2012) Immunopathology, diagnosis, and management of hypersensitivity pneumonitis. Semin Respir Crit Care Med 33(5): 543- 554.
-
Churg A, Muller NL, Flint J, Wright JL (2006) Chronic hypersensitivity pneumonitis. Am J Surg Pathol 30(2): 201-208.
-
Sinha AK (2019) Hypersensitivity Pneumonitis in North Indian population. European Respiratory Journal 54(63S).
-
Pereira CAC, Gimenez A, Kuranishi L, Storrer K (2016) Chronic hypersensitivity pneumonitis. J Asthma Allergy 9: 171-181.
-
Hyldgaard C, Hilberg O, Muller A, Bendstrup E (2014) A cohort study of interstitial lung diseases in central Denmark. Respir Med 108(5): 793-799.
-
Dodaran MS, West J, Smith C, Hubbard R (2007) Extrinsic allergic alveolitis: incidence and mortality in the general population. QJM an International Journal of Medicine 100(4): 233-237.
-
Singh S, Collins BF, Sharma BB, Joshi JM, Talwar D, et al. (2017) Interstitial lung disease in India: results of a prospective registry. Am J Respir Crit Care Med 195(6): 801-813.
-
Hanak V, Golbin JM, Ryu JH (2007) Causes and presenting features in 85 consecutive patients with hypersensitivity pneumonitis. Mayo Clin Proc 82(7): 812-816.
-
Lynch DA (2019) CT Phenotypes in Hypersensitivity Pneumonitis. Chest 155(4): 655-656.
-
Cramer C, Schlünssen V, Bendstrup E, Stokholm ZA, Vestergaard JM, et al. (2016) Risk of hypersensitivity pneumonitis and interstitial lung diseases among pigeon breeders. Eur Respir J 48(3): 818-825.
-
Valencia RF, Camarena A, Pineda CL, Granadoset J, Vega A, et al. (2014) Genetic susceptibility to multicase hypersensitivity pneumonitis is associated with the TNF-238 GG genotype of the promoter region and HLA- DRB1*04 bearing HLA haplotypes. Respir Med 108(1): 211-217.
-
Cardoso J, Carvalho I (2014) The value of family history in the diagnosis of hypersensitivity pneumonitis in children. J Bras Pneumol 40(2): 183-187.
-
Miller R, Allen TC, Barrios RJ, Beasley MB, Burke L, et al. (2018) Hypersensitivity Pneumonitis A Perspective From Members of the Pulmonary Pathology Society. Arch Pathol Lab Med 142(1): 120-126.
-
Vasakova M, Morell F, Walsh S, Leslie K, Raghu G (2017) Hypersensitivity pneumonitis: perspectives in diagnosis and management. Am J Respir Crit Care Med 196(6): 680-689.
-
Selman M, Pardo A, King TE (2012) Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 186(4): 314-324.
-
Vogelmeier C, Krombach F, Munzing S, Konig G, Mazur G, et al. (1993) Activation of blood neutrophils in acute episodes of farmer’s lung. Am Rev Respir Dis 148(2): 396-400.
-
Salinas M, Fritz R, Meneses M, Diaz JC, Rodriguez JC, et al. (2013) Histopathological findings of biopsies from patients with extrinsic allergic alveolitis (hypersensitivity pneumonia) in subacute or chronic phase. European Respiratory Journal 42 (57S).
-
Myers J (2012) Hypersensitivity pneumonia: the role of lung biopsy in diagnosis and management. Mod Pathol 25: S58-S67.
-
Fink JN (1992) Hypersensitivity pneumonitis. Clin Chest Med 13(2): 303-309.
-
Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, et al. (2012) An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med 185(9):1004-1014.
-
Picchio V, Cammisotto V, Pagano F, Carnevale R, Chimenti I (2020) Cell Interaction: Regulation of Immune Responses. Disease Development and Management Strategies, pp: 1-15.
-
Raghu G, Jardin MR, Ryerson CJ, Myers JL, Kreuter M, et al. (2020) Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine 202(3): e36-e69.
-
Vogelmeier C, Krombach F, Munzing S, Mazur G, Beinert T, et al. (1993) Activation of blood neutrophils in acute episodes of farmer’s lung. Am Rev Respir Dis 148(2): 396-400.
-
Akashi T, Takemura T, Ando N, Eishi Y, Kitagawa M, et al. (2009) Histopathologic analysis of sixteen autopsy cases of chronic hypersensitivity pneumonitis and comparison with idiopathic pulmonary fibrosis/usual interstitial pneumonia. Am J Clin Pathol 131(3): 405-415.
-
Salinas M, Fritz R, Meneses M, Diaz JC, Rodriguez JC, et al. (2013) Histopathological findings of biopsies from patients with extrinsic allergic alveolitis (hipersensitivity pneumonia) in subacute or chronic phase. European Respiratory Journal 42(57S).
-
Coleman A, Colby TV (1988) Histologic diagnosis of extrinsic allergic alveolitis. Am J Surg Pathol 12(7): 514- 518.
-
Hariri LP, Kenudson MM, Shea B, Onozato M, Yagi Y, et al. (2012) Distinct histopathology of acute onset or abrupt exacerbation of hypersensitivity pneumonitis. Hum Pathol 43(5): 660-668.
-
Jer TH, Henry TS, Veeraraghavan S, Pardeep KM, Brent PL (2017) Pulmonary Function Tests for the Radiologist. RadioGraphics 37(4): 1037-1058.
-
Boccabella C, Macaluso C, Kokosi M, Alfieri V, Stock C, et al. (2018) Pulmonary function trends predict mortality in patients with hypersensitivity pneumonitis European Respiratory Journal 52(62 S).
-
Santana PV, Prina E, Albuquerque ALP, Carvalho CRR, Caruso P (2016) Identifying decreased diaphragmatic mobility and diaphragm thickening in interstitial lung disease: The utility of ultrasound imaging. Jornal Brasileiro de Pneumologia 42(2): 88-94.
-
Boccatonda A, Decorato V, Cocco G, Marinari S, Schiavone C (2018) Ultrasound evaluation of diaphragmatic mobility in patients with idiopathic lung fibrosis: A pilot study. Multidisciplinary Respiratory Medicine 14(1): 10- 15.
-
Hirschmann JV, Pipavath SNJ, Godwin JD (2009) Hypersensitivity Pneumonitis: A Historical, Clinical, and Radiologic Review. Radiographics 29(7): 1921-1938.
-
Lima MS, Coletta EN, Ferreira RG, Jasinowodolinski D, Arakaki JS, et al. (2009) Subacute and chronic hypersensitivity pneumonitis: histopathological patterns and survival. Respir Med 103(4): 508-515.
-
Silva CI, Muller NL, Lynch DA, Everett DC, Brown KK, et al. (2008) Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin section CT. Radiology 246(1): 288-297.
-
Kligerman SJ, Henry T, Lin CT, Franks TJ, Galvin JR (2015) Mosaic Attenuation: Etiology, Methods of Differentiation, and Pitfalls. Radiographics 35(5): 1360-1380.
-
Tarry D, Powell M (2017) Hypoxic pulmonary vasoconstriction. BJA Education 17(6): 208-213.
-
Zompatori M, Calabrò E, Chetta A, Chiari G, Marangio E, et al. (2003) Chronic hypersensitivity pneumonitis or idiopathic pulmonary fibrosis? Diagnostic role of high resolution Computed Tomography (HRCT). Radiol Med 106(3): 135-146.
-
Montner SM, Husain A, Adegunsoye A, Rekha V, Chung JH (2016) Imaging of Hypersensitivity Pneumonitis. Radiol Clin North Am 54(6): 1033-1046.
-
Chung JH, Zhan X, Cao M, Koelsch TL, Manjarres DCG, et al. (2017) Presence of Air Trapping and Mosaic Attenuation on Chest Computed Tomography Predicts Survival in Chronic Hypersensitivity Pneumonitis. Ann Am Thorac Soc 14(10): 1533-1538.
-
Barnett J, Molyneaux PL, Maher TM, Wells AU, Devaraj A, et al. (2019) Variable Utility of Mosaic Attenuation to Distinguish Fibrotic Hypersensitivity Pneumonitis from Idiopathic Pulmonary Fibrosis. European Respiratory Journal 60(6).
-
S Silva CI, Churg A, Müller NL (2007) Hypersensitivity Pneumonitis: Spectrum of High-Resolution CT and Pathologic Findings. American Journal of Roentgenology 188(2): 334-344.
-
Baldi BG, Carvalho CRR, Dias OM, Marchiori E, Hochhegger B (2017) Diffuse cystic lung diseases: differential diagnosis. J Bras Pneumol 43(2): 140-149.
-
Franquet T, Hansell DM, Senbanjo T, Jardin MR, Muller NL (2003) Lung cysts in subacute hypersensitivity pneumonitis. J Comput Assist Tomogr 27(4): 475-478.
-
Lalancette M, Carrier G, Laviolette M, Ferland S, Rodrique J, et al. (1993) Farmer’s lung: long-term outcome and lack of predictive value of bronchoalveolar lavage fibrosing factors. Am Rev Respir Dis 148(1): 216-221.
-
Cormier Y, Brown M, Worthy S, Racine G, Muller NL (2000) High-resolution computed tomographic characteristics in acute farmer’s lung and in its follow-up. Eur Respir J 16(1): 56-60.
-
Baqir M, White D, Ryu JH (2018) Emphysematous changes in hypersensitivity pneumonitis: A retrospective analysis of 12 patients. Respir Med Case Rep 24: 25-29.
-
Tornyos A, Trinker M, Foris V, Pfeiffer S, Odler B, et al. (2016) Pulmonary hypertension in hypersensitivity pneumonitis. European Respiratory Journal 48(60S).
-
Zangiabadi A, De Pasquale CG, Sajkov D (2014) Pulmonary Hypertension and Right Heart Dysfunction in Chronic Lung Disease. BioMed Research International 2014: 1-13.
-
Nathan SD (2014) Hypersensitivity pneumonitis and pulmonary hypertension: How the breeze affects the squeeze. European Respiratory Journal 44(2): 287-288.
-
Oliveira RKF, Pereira CAC, Ramos RP, Ferreira EVM, Messina CMS, et al. (2014) A haemodynamic study of pulmonary hypertension in chronic hypersensitivity pneumonitis. European Respiratory Journal 44(2): 415- 424.
-
Remy-Jardin M, Ryerson CJ, Schiebler ML, Leung ANC, Wild JM, et al. (2021) Imaging of Pulmonary Hypertension in Adults: A Position Paper from the Fleischner Society. Radiology 298(3): 531-549.
-
Walsh SLF (2019) Mediastinal Lymphadenopathy in Interstitial Lung Disease. Time to Be Counted. American Journal of Respiratory and Critical Care Medicine 199(6): 685-687.
-
Küpeli E, Karnak D, Kayacan O, Beder S (2004) Clues for the differential diagnosis of hypersensitivity pneumonitis as an expectant variant of diffuse parenchymal lung disease. Postgrad Med J 80(944): 339-345.
-
Singh S, Collins BF, Sharma BB, Joshi JM, Talwar D, Katiyar S, et al. (2019) Hypersensitivity pneumonitis: Clinical manifestations – Prospective data from the interstitial lung disease-India registry. Lung India 36(6): 476-482.
-
Alvarez AB, Lee AS, Mira-Avendano I (2017) Etiologies of consecutive series of non-necrotizing granulomas. Sarcoidosis Vasc Diffuse Lung Dis 34(2): 115-121.
-
Sadek SH, Khalifa WA, Azoz AM (2017) Pulmonary consequences of hypothyroidism. Ann Thorac Med 12(3): 204-208.
-
Kuramochi J, Inase N, Miyazaki Y, Kawachi H, Takemura T, et al. (2011) Lung cancer in chronic hypersensitivity pneumonitis. Respiration 82(3):263-267.
-
Kim YJ, Song JW, Kim DS (2019) Acute Exacerbation of Chronic Hypersensitivity Pneumonitis: Incidence, Risk Factors and Outcome. A101 Translational Studies in ILD IPF and Sarcoidosis, American Thoracic Society International Conference, pp: A2418-A2418.
-
Pérez-Padilla R, Salas J, Chapela R, Sánchez M, Carrillo G, et al. (1993) Mortality in Mexican patients with chronic pigeon breeder’s lung compared with those with usual interstitial pneumonia. Am Rev Respir Dis 148(1): 49- 53.
-
Walsh SL, Sverzellati N, Devaraj A, Wells AU, Hansell DM (2012) Chronic hypersensitivitypneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur Radiol 22(8): 1672-1679.
-
Salisbury ML, Gu T, Murray S, Gross BH, Chughtai A, et al. (2019) Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest 155(4): 699- 711.
-
Creamer AW, Barratt SL (2020) Prognostic factors in chronic hypersensitivity pneumonitis. European Respiratory Review 29(156): 190167.
-
Adegunsoye A, Oldham JM, Demchuk C, Montner S, Vij R, et al. (2016) Predictors of survival in coexistent hypersensitivity pneumonitis with autoimmune features. Respir Med 114: 53-60.
-
Pardo A, Barrios R, Gaxiola M, Segura-Valdez L, Carrillo G, et al. (2000) Increase of lung neutrophils in hypersensitivity pneumonitis is associated with lung fibrosis. Am J Respir Crit Care Med 161(5): 1698-1704.
-
Blanchet MR, Israël-Assayag E, Cormier Y (2004) Inhibitory effect of nicotine on experimental hypersensitivity pneumonitis in vivo and in vitro. American Journal of Respiratory and Critical Care Medicine 169(8): 903-909.
-
Wälscher J, Gross B, Morisset J, Johannson KA, Vasakova M, et al. (2020) Comorbidities and survival in patients with chronic hypersensitivity pneumonitis. Respiratory Research 21(1): 12.
-
Rittig AH, Hilberg O, Ibsen R, Løkke A (2019) Incidence, comorbidity and survival rate of hypersensitivity pneumonitis: a national population-based study. ERJ Open Research 5(4): 00259-02018.
- Ultrasound Guided Therapeutic Nerve Blocks
- Cyclops Lesion Without ACL Reconstruction: A Rare Case in a Patient with Intact Anterior Cruciate Ligament and Tibial Plateau Fracture
- Dosimetric Comparison between Two Dose Calculation Algorithms in SBRT Treatment of Lung Cancer in Ring-based and C-arm Radiation Therapy Equipment
- Adolescent Testicular Adrenal Rest Tumors: A Case Report and Review of the Literature
- Giant Intrathoracic Lipoma: A Rare Presentation
- Image of a Right Renal Angiomyolipoma Complicated by Hemorrhage