Targeting Redox Enzymes of Cholesterol Biosynthesis: A Step in the Right Direction Towards Ending Cardiovascular Disease
Global occurrence of cardiovascular diseases is a serious health concern resulting in over 2 million deaths annually. A major cause of cardiovascular disease is elevated levels of blood plasma cholesterol which leads to the precipitation of lipoproteins and arterial plaque formation. This review details treatment of hypercholesterolemia that target redox enzymes involved in the biosynthesis of the lipid. It begins with a brief discussion of both high- and low-density lipoproteins that are the vehicles for cholesterol transport in humans. An overview of the five stages of cholesterol anabolism in the liver is then provided focusing on the two rate-limiting redox enzymes involved in the pathway. These enzymes are 3-hydroxy-3-methylglutyl CoA reductase (HMGR) and squalene monooxygenase (SMO). The structure and catalytic mechanisms are presented in great detail to lay the foundation for a presentation of common cholesterol lowering drugs. The first of these are the statins that competitively inhibit HMGR and have shown the greatest success in both the treatment and prevention of cardiovascular diseases. An inhibitor of SMO called terbinafine is then discussed. This drug molecule is a fairly effective anti-fungal but is not currently approved in the U.S. to treat high cholesterol. However, it has been shown to lower low-density lipoproteins and exhibits promise to provide the framework for the development of new cholesterol lowering treatments to target SMO. It is hoped that this review article will raise awareness and spur future research into the treatment of hypercholesterolemia to treat and/or prevent cardiovascular disease.
Introduction
Elevated blood cholesterol is a high-risk factor and one of the leading causes for both the development of cardiovascular disease and the occurrence of strokes. The World Health Organization (WHO) defines “high” as blood plasma concentrations of >5.0 mM (190 mg/dL) of total cholesterol. According to the WHO, a third of global cases of ischemic heart disease is attributed to high cholesterol and this leads to >2.5 million annual deaths [1]. Arteriosclerotic cardiovascular disease remains a problem in countries of all economic levels despite recommendations and goals set by the WHO [2]. In humans, the liver is primarily responsible for generating cholesterol which circulates through the blood stream. Cholesterol production by the liver is intended to repair damaged cells, break down ingested fat, and create hormones for a variety of signing responses. The hydrophobic nature of the lipid leads to aggregation of the molecule and the formation of plaques in patients suffering from hypercholesterolemia [3]. This hinders healthy blood flow from the vessels to the coronary arteries preventing the supply of oxygen to tissues. This causes to cell death, and if plaque formation over time causes a complete blockage of blood flow, cardiac arrest occurs.
![Figure 1: General Lipoprotein Structure. The hydrophilic surface allows the particle to be freely transported throughout the blood stream. Cholesterol attaches to protein through ester linkages that are cleaved by phospholipases when the lipid is needed. This figured was modified from reference [4].](/fulltextimages/10679/fig_1.png)
As shown in Figure 1, cholesterol largely contains non-polar covalent bonds between carbon and hydrogen atoms. It therefore has limited solubility in the aqueous environment of blood plasma. Thus, cholesterol does not freely circulate throughout the blood stream, but rather is associated with small spherical proteins called lipoproteins. A detailed account of lipoproteins is beyond the scope of this review, so interested readers are a referred to [4] and the references therein. Briefly, cholesterol is transported by the two physically smallest of the seven general classes of lipoproteins. These two classes are the low- and high- density lipoproteins (LDL and HDL, respectively) and have particle sizes of between only 5-30 nm in diameter [4]. HDLs are commonly referred to as “good” cholesterol and LDLs as “bad” though these terms are misnomers because the sterol molecules associated with the proteins are identical in each case.
The general structure of lipoproteins is shown in Figure 1. They resemble micelles and contain a hydrophilic surface to increase blood plasma solubility and a hydrophobic core where the bulk of cholesterol molecules are transported. Cholesterol attaches to lipoproteins mainly as ester linkages to the proteogenic core. LDLs hold the majority of cholesterol transported through humans and most consist of a 512 kDa protein core called Apo B-100 [5]. Accumulation of LDLs in the blood stream leads to sterol deposits, plaques and the risk of cholesterol molecules breaking off into segments. Water insoluble pieces form clots, which will physically block off access of blood to the arteries. This can result in pulmonary embolism or venial thrombosis [6]. In order to heal and repair damaged cells, LDLs collectively embed within the vessels of the heart. Phospholipase C (E.C. 3.1.4.11) will liberate cholesterol molecules from LDLs when needed by catalyzing ester hydrolysis to cleave the covalent bond linking it to the protein [7].
HDLs are generally larger than LDLs and play an important role in transporting cholesterol away from peripheral tissues and back to the liver for expulsion from the body [4]. HDLs are more tightly packed than LDLs and consist of multiple core apoproteins ranging between 6.6-34 kDa [4]. Tight packing of the apoproteins give particle sizes of between 5-10 nm in diameter [8]. As their name implies, they contain more cholesterol molecules per particle. Since their physiological role is to transport cholesterol out of the body, they are generally beneficial to human health [5, 9]. Given the beneficial effects of HDLs, drug treatments specifically designed to increase their level in blood plasma were developed. These include both niacin and fibrate therapies and specific cholesteryl transferase inhibitors such as Anaceptrib® [10]. However, clinical trials were halted since they were shown to have little if any effect on the incidence of heart attacks or cardiovascular disease [11, 12].
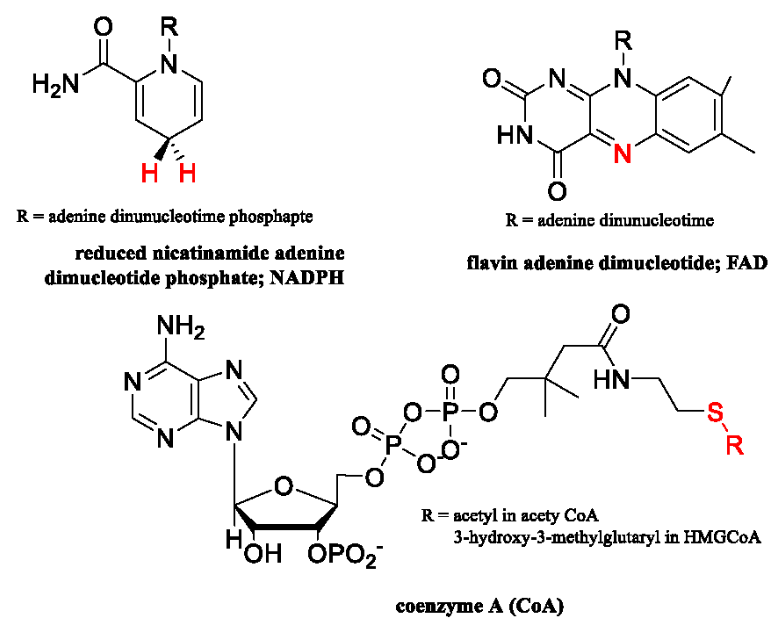
Instead, strategies aimed at inhibiting cholesterol biosynthesis have proven to be quite effective in both treating and preventing cardiovascular diseases. This review will focus on two drugs that target redox enzymes involved in cholesterol production in the liver. We start with an overview of the anabolic pathway of cholesterol biosynthesis in humans. The role of statins such as Lipitor® and Crestor® in lowering LDL levels will then be presented through a detailed presentation of the structure and inhibition of 3-hydroxy-3-methylglutryl-CoA reductase (HMGR). This enzyme is involved in the mevalonate pathway that contributes to cholesterol production in hepatocytic cells. We then move onto a similar discussion of the drug terbinafine (brand name Lamisil®). This drug was developed to treat fungal infections, but has been demonstrated to also lower cholesterol in humans. The drug targets squalene monooxygenase (SMO) which is another redox enzyme involved in cholesterol biosynthesis. Detailed descriptions of the structure and inhibition of SMO by the drug will be outlined. Before we start our discussion, structures of the coenzymes used by HMGR and SMO are presented in Figure 2 since they may not be familiar to all readers. We hope this review is informative and may stimulate further research in the treatment and prevention of cardiovascular diseases.
Chlosterol Biosynthesis in Liver and Instestine
The liver is primarily responsible for maintaining homeostatic blood plasma cholesterol levels [13]. Roughly 75-80% of cholesterol in humans is synthesized using acetyl- CoA precursors while the other 20-25% is derived from dietary consumption of animal products. New cholesterol molecules are produced in hepatic cells for transport by LDLs. These lipid molecules are transferred back to the liver by HDLs where they are converted to bile acids and excreted by the digestive system [14]. Dietary cholesterol is absorbed by the small intestine, incorporated into lipoprotein particles called chylomicrons and transported to the liver for incorporation into LDLs [15]. Given the crucial physiological role of cholesterol, nearly all cell types can synthesize the lipid. However, the predominant pathway of cholesterol biosynthesis takes place in hepatic cells starting from acetyl- CoA units produced during catabolism of fats, proteins and carbohydrates.
![Figure 3: Structure of HMGR dimer. The structure shown is for the enzyme from P. mevalonii bound to a tetrahedral dithiohemiacetal intermediate (PDB ID: 1QAX) and shows the active site residues which are conserved in all HMGRS. The figure was modified from [28].](/fulltextimages/10679/fig_3.png)
Scheme 1: Overview of Cholesterol Biosynthesis Cholesterol is produced from acetate in five stages. Statins target stage one whereas terbinafine targets stage four. Stereochemical configurations are omitted from the structures for simplicity. The inset shows the structure HMGR which statins competitively inhibits to block cholesterol production.
Scheme 1 shows a simplified illustration of this pathway that highlights the stages targeted by cholesterol lowering drugs. Elucidation of the metabolic pathway for cholesterol biosynthesis was pioneered by Bloch [16], Kundutsch and Russel [17] and others. Early experiments were conducted by Konrad Bloch who gained considerable experience in isotopic labeling experiments in the late 1930s and early 1940s as a graduate student at Columbia University. The technology was new at the time and his early projects involved the platinum catalyzed deuteriation of alkenes to synthesize 2H-labeled biomolecules as metabolic tracers. These isotopically labeled molecules effectively mapped metabolic processes under different physiological conditions [18]. Bloch later moved to the University of Chicago and set out to determine where all 27 carbon atoms of cholesterol and other sterols are derived. His work on this topic spanned a couple of decades and led to not only a detailed understanding of cholesterol anabolism, but the identity of enzyme targets for treatment of cardiovascular diseases.
Early experiments started with 13C-labeled acetate fed to pyruvate-deficient mutants of Neurospora crassa. This led to the isolation of labeled ergosterol [19] and the hypothesis of a pathway involving acetate being converted into a cyclized intermediate before transforming into cholesterol. In the 1950s, Bloch and fellow researcher, R. G. Langdon, demonstrated that acetate is converted into squalene through a total of 11 reactions [20]. Through additional 13C labeling studies, Bloch was then able to pinpoint the origin of the hydroxyl groups found in sterols and to establish it is critical to convert squalene to cholesterol [19]. In 1960, Kundutsch and Russel also used isotopic labeling studies to discover an alternate pathway that coverts squalene to cholesterol. Their pathway differs in only a few small details [17]. The Bloch and Kundutsch-Russel pathways do not contradict each other but rather reflect the evolution of cellular flexibility to adapt to changing metabolic demands under different physiological conditions [13].
Cholesterol production can be divided into five general stages and involves 24 enzymatic reactions. The first stage is referred to as the mevalonate or isoprenoid pathway and, in the context of cholesterol anabolism, involves three reactions (mevalonate is also a precursor for many vitamins, coenzymes and steroids) [21]. All carbon atoms fed into stage one are derived from acetate and carried into the cell by acetyl-CoA. Importantly, the last reaction of stage one involving the reduction of 3-hydroxy-3-methylglutyl coenzyme A (HMG-CoA) and is effectively targeted by statins to reduce human LDL levels as discussed below. Stage two involves five reactions and converts mevalonate to activate isoprenoids [21]. The next stage links these isoprenoids to generate an unsaturated hydrocarbon of 30 carbon atoms known as squalene [20, 22]. In stage four, squalene undergoes three reactions to form a fused-cyclic molecule called lanosterol. The first of these reactions is also targeted by another drug, terbinafine [23, 24]. Lanosterol is converted to cholesterol in stage five by either the Bloch or Kundutsch- Russel pathway. Both of these occur in ten enzyme catalyzed reactions [16, 17, 19].
Targeting enzymes involved in the metabolic pathway illustrated in Scheme 1 coupled with diet and exercise effectively lowers the risk of both cardiovascular diseases and stroke [25]. Blocking the metabolic production of cholesterol significantly lowers blood plasma LDL levels since it limits the number of molecules to load onto proteins. This stimulates the liver to produce both phospholipases and bile acids that degrade LDLs for expulsion from the body through the digestive tract [7, 10]. It also leads to increased levels of LDL receptors on hepatic cells that take up LDLs and reduce their circulation in the blood stratum [26]. The sections below discuss the targeting of two redox enzymes involved in cholesterol anabolism; namely HMG-CoA reductase (HMGR; E.C. 1.1.1.34) and squalene monooxygenase (SMO; E.C. 14.13.132). Before starting this discussion, we shall outline the current knowledge of the structural and mechanistic properties of these enzymes.
Structure and Mechanism of HMGR and SMO
Enzyme Target of Statins: HMGR catalyzes the rate controlling reaction of the mevalonate pathway as shown in Scheme 2. This reaction involves the stereoselective reduction of the thioester bond of HMG-CoA by NAD (P) H to generate (R)-mevalonate.1 Owing to the critical role of (R)-mevalonate in not only cholesterol production but a plethora of other metabolites, HMGR has been extensively studied for almost a century. The enzyme from the eubacteria Pseudomonas mevalonii has been most thoroughly characterized since it is cytosolic and therefore, easy to work with. It was found to be overexpressed simply by growing the bacterium in media containing mevalonate as the sole carbon source [27] and is also readily purified to high levels [28]. The first crystal structure of HMGR was solved in 1995 [29]. Many others have been solved since from a variety of sources [21, 28, 30, 31]. Eukaryotic HMGRs are membrane bound so contain both a transmembrane domain and a catalytic domain. They only share 14-20% overall sequence similarity with prokaryotic HMGRs, but have several conserved active site residues. They are also proposed to utilize the same general catalytic mechanism based on numerous kinetic, structural and computational studies [28, 31].
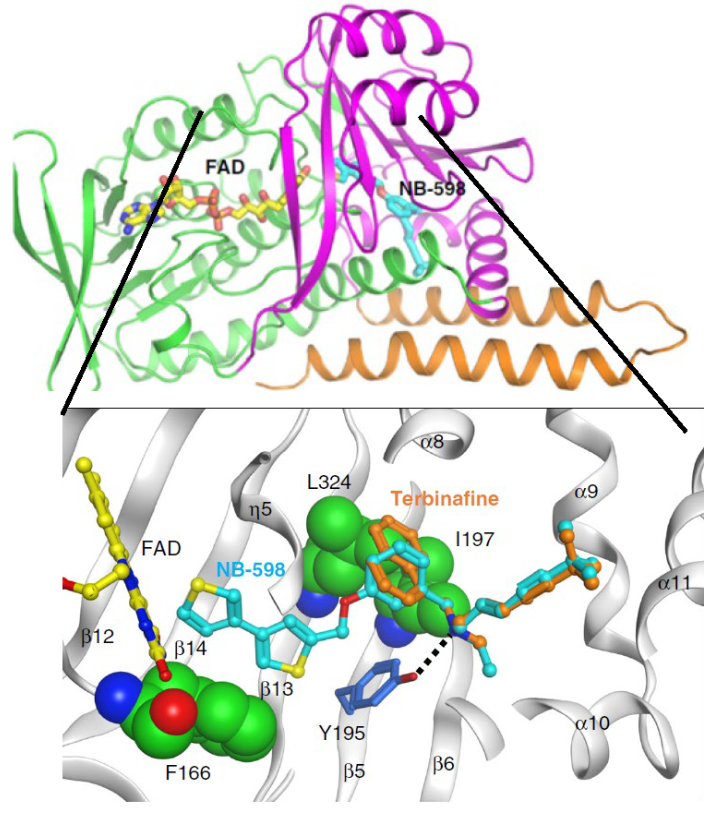
![Figure 4: Structure of Human SMO and Computational Model of SMO-Terbinafine Complex. The structure of SMO was solved in complex with the small molecule inhibitor NB-598. This structure was used to create a model of the enzyme with the anti- fungal drug terbinafine. The figure was modified from Padyana, et al. [24].](/fulltextimages/10679/fig_4.png)
Scheme 2: Reaction Catalyzed by 3-Hydroxy-3-methylglutaryl CoA Reductase (HMGR). Structures of NADPH and CoA are shown in Figure 1.
Human HMGR is a tetramer of two obligate dimers that form quite similarly to the dimeric structure of P. mevalonii enzyme [28]. Figure 3 shows the general structure of the HMGR dimer as well as the conserved residues in of the active site of the enzyme. A so-called, “flap domain” runs along one side of the active site to position a catalytic lysine residue in proximity of HMG-CoA (Lys 267 in Figure 3) [32]. Closing of the flap positions other catalytic residues as illustrated in Figure 3. The carboxylate of Asp767 in the human enzyme is 3.6-3.7 Å away from Glu767 leading Istavan et al to propose that it raises the pKa value of the side chain to such an extent that it can serve as a general acid for the reaction [31]. The equivalent residues in the bacterial enzyme are Asp283 and Glu83. The proposed role of Glu was later confirmed by quantum mechanical/molecular mechanical models of both the bacterial and human HMGR reactions [33]. A final conserved residue that is present in all HMGRs is a histidine (His381 in Figure 3) that acts as a general acid for the cleavage of the thioester bond linking the glutaryl moiety to CoA [28, 29, 31].
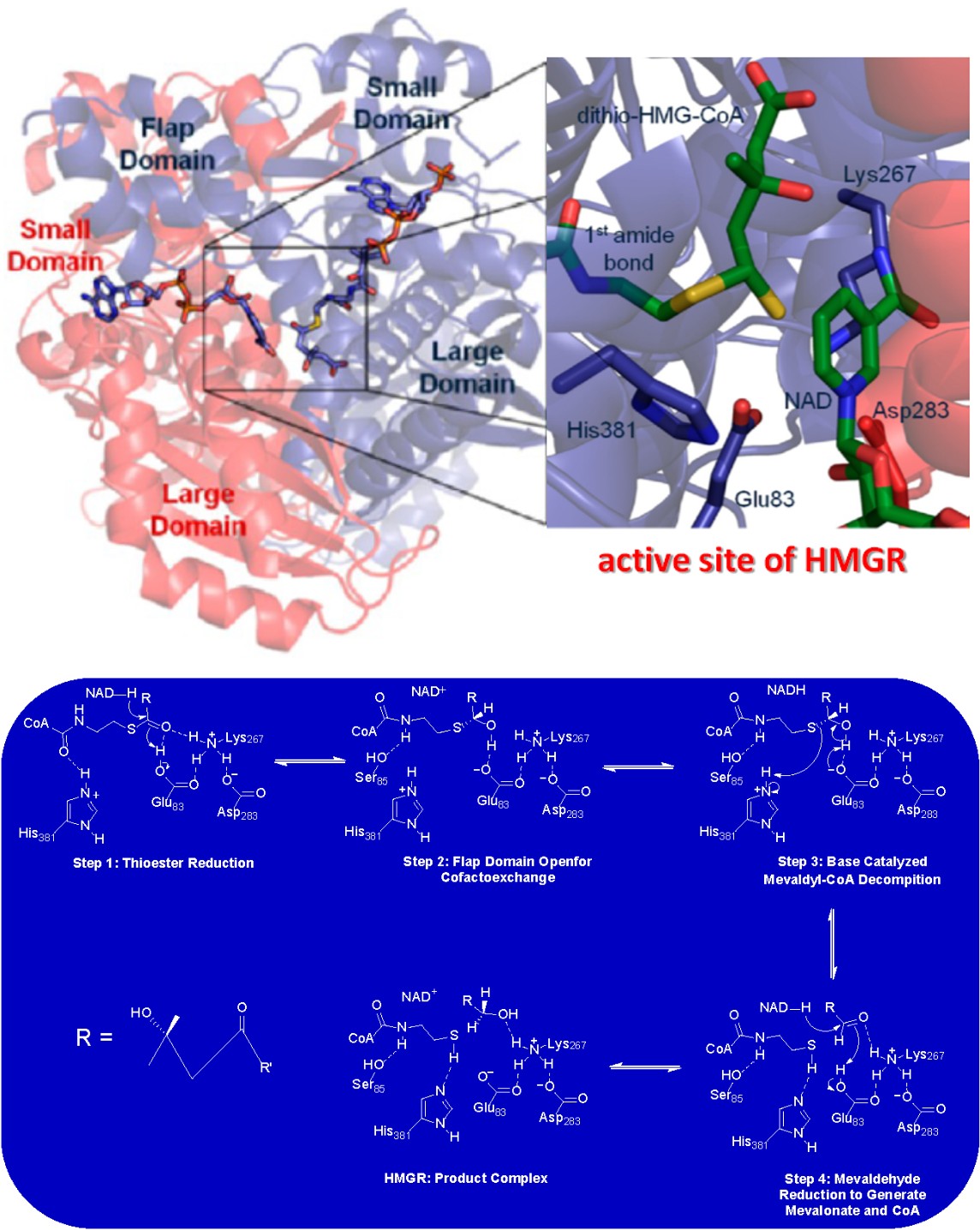
Scheme 3: Catalytic Mechanism of HMGR. The mechanism is proposed based on decades of mechanistic studies [28]. See main text for a description of the chemical details of the reaction.
Elucidation of the complete catalytic mechanism for the HGMR reaction took several decades and was the culmination of many kinetic, structural, computational and molecular biology experiments [28]. These studies spanned over the course of more than 70 years after Bloch’s work reveled that the HMGR reaction controlled the rate of human cholesterol biosynthesis. The generally accepted mechanism proposed for the HMGR is presented in Scheme 3. Residue numbers corresponding to the most extensively studied P. mevalonii enzyme. All available evidence suggests eukaryotic HMGRs, including that from humans, utilize an identical catalytic mechanism [28, 33]. The overall reaction involves a four-electron reduction of HMG-CoA through two separate hydride transfer reactions between the substrate and NAD (P) H. Flap closing in the ternary complex of HGMR initiates catalysis. A hydride ion is first transferred from NAD (P) H to HMG-CoA to reduce the thioester bond to an alcohol (Step 1 in Scheme 3). Reduction occurs with a concomitate proton transfer from Glu83 to the carbonyl oxygen of the thioester of CoA. In the next step of the reaction, the flap domain of HMGR opens to allow for NAD(P)+ to leave the active site and a new reduced cofactor to enter for the next hydride transfer step to occur (Scheme 3, Step 2). Evidence for this step came from both kinetic and structural studies of HMGR with dithio-HMG-CoA as substrate. This analogue was predicted to be a competitive inhibitor for the enzyme but was instead found to be a slow substrate [34]. Structures of the enzyme in complex with dithio-HMG-CoA revealed the conformational changes that occur during cofactor exchange. This involves the formation of a hydrogen bond between a conserved serine (Ser85 in the P. mevalonii enzyme) and the amido hydrogen of CoA. The active site glutamate (Step 3 in Scheme 3) then acts as a general base to abstract the hydroxyl proton of mevaldyl- CoA. This occurs with a concerted sulfide protonation by an active site histidine residue (His381 in Scheme 3). This liberates CoA and leads to a second NAD (P) H dependent reduction to produce the carboxylic acid of mevalonate (Step 4 in Scheme 3).
Enzyme Target of Terfenadine: SMO
SMO catalyzes the stereoselective oxidation of squalene to 2,3(S)-oxidosqualene in the first reaction of Stage 4 for cholesterol biosynthesis (as outlined in Scheme 1) [3, 20, 24]. This reaction is the second slowest step in the metabolic pathway for cholesterol biosynthesis, and like HMGR, has drawn considerable attention by both biochemists and structural biologists alike. As shown in Scheme 4, SMO catalyzes the oxidation of squalene through the incorporation of a single oxygen atom from molecular oxygen to the substrate with the production of water as a byproduct. Since the enzyme produces an epoxide it is also often referred to as squalene epoxidase, but will only be referred to as a monooxygenase in this review article to conform to STRENDA nomenclature rules. SMO is a flavin adenine dinucleotide (FAD) dependent enzyme that utilizes NADPH as coenzyme [35]. It is classified as a Group E flavin monooxygenase based on sequence and structural homology to other enzymes in this superfamily [24, 36, 37]. While it has yet to be rigorously experimentally validated, it is reasonable to assume that SMO utilizes a similar catalytic mechanism as the paradigm Group E flavin monooxygenase, styrene monooxygenase (E.C. 1.14.14.11), as discussed below.
![Figure 6: Inhibition of SMO by Terbinafine. The data was taken from Padyana AK, et al. [24] which showed that terbinafine inhibits human SMO with a IC50 value of ~7 mM. The structure of terbinafine is shown with a box of the enzyme moiety that was shown to be critical for binding for this and other SMO inhibitors.](/fulltextimages/10679/fig_6.png)
Scheme 4: Reaction Catalyzed by Squalene Monooxygenase (SMO). The oxygen atom incorporated into the product is highlighted in red.
In 2019 Padyana, et al. solved the structures of human SMO in either the absence or presence of the small molecule inhibitors NB-598 and Cmpd-4’’ [24]. Free enzyme was solved to a resolution of 3.0 Å, ligand bound structures were resolved to 2.3-2.5 Å and comparison of the two provided many insights into inhibition by terbinafine (see TERBINIFINE INHIBITS OF SMO ALSO LOEWERS LDL LEVELS IN HUMAN AND FUNGI below). The overall structure of the enzyme (PDB ID: 6C6P) in complex with NB-190 is shown in Figure 3 and is presented here to focus on its classification as a Group E flavin monooxygenase (interested readers should consult Padyana, et al. [24] for the structure of NB-190 which is omitted here for simplicity). Proper classification of SMO has important mechanistic implications that have been difficult to experimentally verify due to the laborious techniques required to kinetically characterize the enzyme [24]. Early computational models of SMO were developed using p-hydroxybenzoate hydroxylase (E.C. 1.14.13.2), a Group A FMO, as the starting point [23]. This was based on sequence analysis showing that SMO has conserved residues for a Rosman fold to bind FAD and a similar tertiary structure as p-hydroxybenzoate hydroxylase. Advances in the field of flavin monooxygenase enzymology since 2010 [36, 37], along with the long-held demonstration that the enzyme requires an external electron donor for activity [38, 39] argues against SMO being Group A enzyme. Instead, mounting evidence suggests it is a Group E enzyme [36, 37]. SMOs contain many conserved aromatic amino acids residues that are essential to catalysis [40] and several were found in the active site of the enzyme [24].
Unlike Group A enzymes, the flavin cofactor of SMO does not remain bound throughout catalytic turnover, but instead is provided by an external reductase [36, 37]. Molecular biology experiments have demonstrated that the electrons used to reduce the FAD coenzyme of SMO are provided by a NAD (P) H dependent cytochrome P450 reductase [38, 39] and these experiments were confirmed in biochemical studies of the enzyme [24]. Reduced FAD is transferred to SMO for the epoxidation reaction. The requirement of an external reductase coupled with the formation of an epoxide from an alkene strongly supports the classification of SMO as a Group E flavin monooxygenase. Styrene monooxygenase (E.C. 1.14.14.11) [41, 42] and zeaxanthin epoxidase (E.C. 1.14.13.90) [43] are the only two other members of Group E. They catalyze the same type of reaction using NAD (P) H as reductant.
Sophisticated liquid choreography/mass spectrometric based assays were developed in [24] for kinetic assays of recombinant SMO overexpressed in baculosomes. This laborious methodology has prevented detailed mechanistic validation of SMO, but given its similarities to other Group E flavin monooxygenases, it is reasonable to assume it utilizes the catalytic mechanism. Scheme 5 proposes such a mechanism for the first time based on extensive rapid kinetic data collected for styrene monooxygenase which rigorously characterized the redox states of FAD during substrate oxidation [42]. SMO catalysis requires FAD reduction by cytochrome P450 reductase (step 1 in Scheme 5) as shown in [39]. Once reduced FAD is transferred to SMO (Scheme 5; step 2) squalene epoxidation can commence. The crystal structure of human SMO shows that the N(5) position of the isoalloxazine ring of FADH2 is stabilized through hydrogen bonding with Tyr335. Step 3 would involve atmospheric oxygen reacting with the SMO: FADH2 binary complex to produce the peroxyflavin form of the coenzyme. Stabilization of a peroxyflavin was clearly observed for styrene monooxygenase through rapid mixing of the reduced enzyme with oxygen. Time resolved flavin spectra showed the characteristic peak of peroxyflavins centered at ~380 nm from 8-100 ms after mixing [42]. Such an intermediate in SMO would be stabilized by the active site residue Tyr195 (Scheme 5; step 3). The same residue would act as a general acid to protonate the peroxyflavin to form the hyroperoxyflavin form of FAD (Scheme 5; step 4). In step 5, squalene would bind to the enzyme for rapid epoxidation (that was too fact to detect with styrene monooxygenase [42]). The nucleophile required to initiate the epoxidation would be provided by the p-electrons of the C4 carbonyl bond of the isoalloxazine ring of the flavin. A concerted reaction following a well-established mechanism [44] between the 2,3 alkene of squalene and the hydroperoxyl group of FAD would then take place as shown in Step 6 of Scheme 5. The isoalloxazine C4 carbonyl would be regenerated through a Tyr195 catalyzed proton abstraction. This would take place with the concomitant expulsion of water from the hyroperoxyflavin a well-documented process with flavin monooxygenases [36, 37].
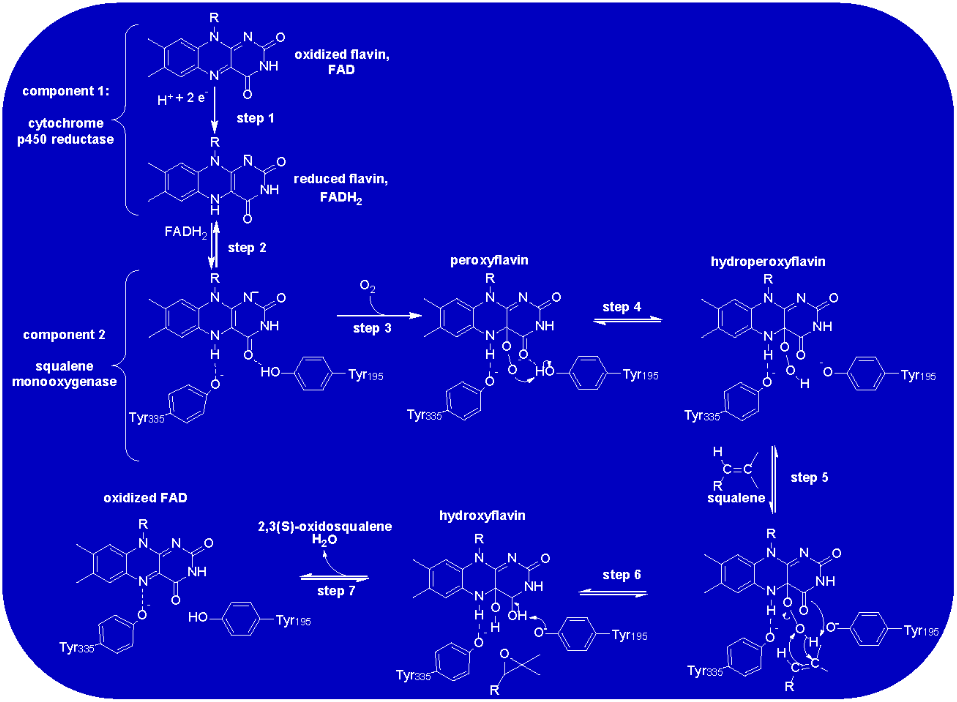
Scheme 5: Proposed Catalytic Mechanism of SMO. The mechanism is proposed for the first time in this study based on both extensive mechanistic data collected for styrene monooxygenase [42] and the structural studies of human SMO [24]. Further studies are required to validate this proposal.
Statins Lower LDL levels through Inhibition of HMGR to Prevent Cholesterol Synthesis
Konrad Bloch and Feodor Lynch were awarded the 1964 Nobel Prize in Physiology or Medicine for their work on cholesterol biosynthesis [18]. Their work ushered in new areas of study for the generation of safe and effective cholesterol lowering drugs to treat cardiovascular diseases [45, 46]. It was quickly realized that HMGR should be a prime target since it catalyzes the committed step of cholesterol biosynthesis and therefore is naturally regulated in human metabolism to maintain cholesterol homeostasis [15]. The earliest statin ever developed to treat hypercholesterolemia was discovered by the Japanese pharmaceutical company Sankyo (now Dalichi Sankyo Limited) in 1977 [47]. This statin was isolated from the blue-green mold Penicillium citrinum and has come to be known as compactin (structure shown in Figure 5). While animal studies in chickens, dogs and monkeys all showed promising cholesterol lowering activity [48], Sankyo discontinued its development in 1980 after the discovery of lymphomas in dogs treated with the drug [49]. Around the same time, Merck, inspired by the compactin studies, isolated another statin from the soil fungus Aspergillus terreus [50]. The same statin was also identified from the purple mold Monascus ruber and was found to be quite effective in the treatment of hypercholesterolemia without the carcinogenic or other toxic effects observed with compactin [51].
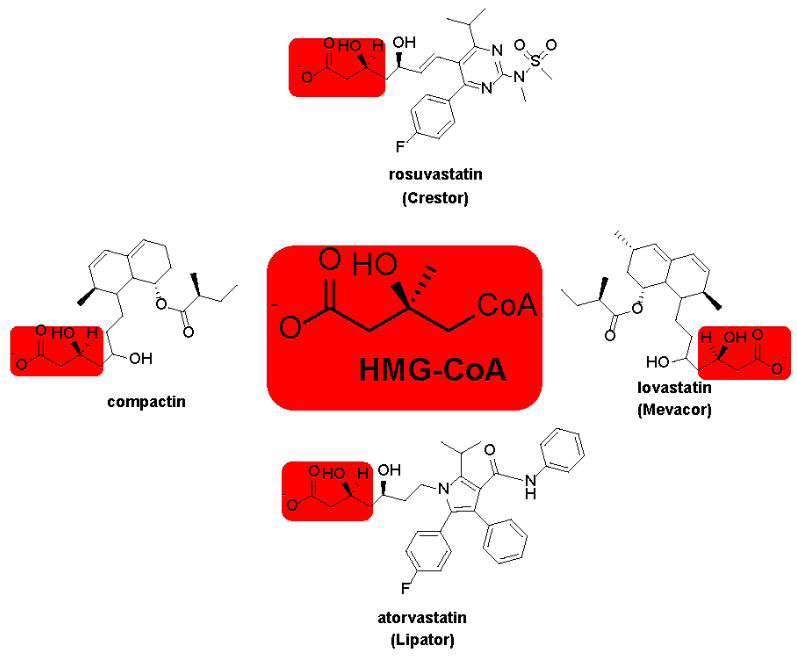
Figure 5: Structure of Some Common Statins. Each is shown in the β-hydroxy form that exists under physiological conditions. Red boxes are to highlight the structural similarities between statins and HMG-CoA, the substrate for HMGR. These similarities give rise to the potent competitive inhibition by statins (Ki in the nM range) to lower cholesterol biosynthesis.
The statin isolated by Merck has come to be known as lovastatin and was carried through clinical trials in the 1980s. Structural similarities between compactin, lovastatin and HMG-CoA (Figure 5) makes it no surprise that statins are potent competitive inhibitors of HMGR [47, 50]. As with the HMG-CoA, the carboxylate oxygen of lovastatin (highlighted in red boxes) forms a lactone with the γ-carbon of the drug, but this readily undergoes hydrolysis in vivo to give the β-hydroxy acids shown in in Figure 5. Clinical trials were a success and lovastatin got approval from the U.S. Food and Drug Administration in 1987 to be sold under the brand name Mevacor® [49]. Since that time, several statins have been developed and approved for the treatment of high LDL levels. Figure 5 shows a few representative examples and provides both their generic and brand names. Statins have a remarkable potency and competitively inhibit HMGR both in vitro and in vivo with KD or IC50 values in the low nM range [31]. Crystal structures of HMGR: lovastatin complex show that the drug makes four different active site contacts with the enzyme through hydrophobic and hydrogen bonding interactions with the same amino acids used to bind the HMG-Co A substrate [52]. Inhibiting HMGR activity dramatically decreases mevalonate production (Scheme 1) and therefore LDL cholesterol levels. This reduction has been demonstrated to reduce the risk of major cardiovascular events by about 25% after each year of statin therapy [53].
Terbinafine Inhibition of SMO also Lowers LDL levels in Humans and Fungi
As with any drug, statin therapy is sometimes met with intolerable side effects which can lead to a discontinuation of treatment. The most common side effect of statins is muscle pain or weakness, and this often leads to patients not remaining compliant with therapy [54]. In such cases, alternate therapies are desirable and the targeting of SMO by terbinafine could provide such a strategy. Currently, terbinafine is only approved to treat fungal infections typically by a topical ointment, though oral therapies are also available (i.e. Terbinex®). A correlation between terbinafine treatment and lower cholesterol levels have been made in some studies [55, 56, 57]. We end with a look into the inhibition of SMO by terbinafine since it could lead to more effective ways of treating high levels s of LDL in patients suffering from hypercholesterolemia for which statin therapy was unsuccessful.
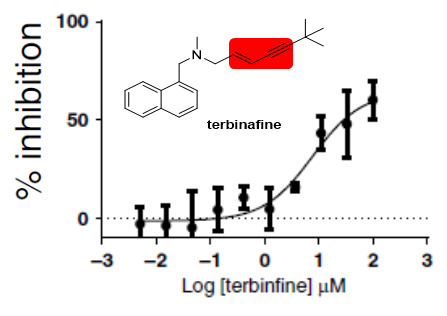
The inhibition of both human and fungal SMO has been well characterized kinetically and structurally [23, 24, 38, 56, 57, 58]. Owing to the inherent difficulties in measuring the kinetics of SMO, the inhibition of the enzyme by terbinafine was not straightforward to characterize. Nevertheless, inhibition is now generally well understood. Fungal SMO is clearly more susceptible to terbinafine inhibition showing KD values in the nM range as compared to the sub-M values for the human enzyme (Figure 6). Comparison of the chemical structures of terbinafine and squalene leads to a reasonable hypothesis that it should competitively inhibit the enzyme. However, kinetic patterns of terbinafine inhibition clearly show that it is a tight binding non-competitive inhibitor [24, 38]. Both computational modeling of terbinafine inhibition of SMO [23] and high-resolution crystal structure of the human enzyme in complex with the structural analogs NB-190 and Cmp-4’’ show many interactions between the drug and active site residues as expected. Non-competitive inhibition patterns could simply be the result of the limited solubility of terbinafine in aqueous solution that prevented substrate displacement.
The inhibition of SMO by terbinafine has similar effects on cholesterol production as does statin inhibition of HMGR. By blocking a rate-controlling step of cholesterol production through enzyme inhibition, cells have nothing to load onto LDLs. This triggers the destruction of LDLs in the liver and has positive clinical outcomes. Inhibition of SMO in fungi prevents cell wall formation and thus survival of the microbe. Weaker terbinafine inhibition of human SMO currently limits its use as a cardioprotective agent. However, the biochemical and structures studies of Padyana, et al. [24] provide a framework on how human SMO inhibitors can be developed to provide new drugs to treat cardiovascular diseases.
Conclusion
Before the work of Konrad Bloch and many other Biochemists in the first half of the 20th century, it was a common belief that cardiovascular events such as heart attacks or strokes were just a normal part of the aging process. It was generally known that high levels of cholesterol in the blood led to plaque formation, but it seemed this could not be avoided. Once the details of cholesterol metabolism were understood, it became apparent that the medical community was not powerless to the aging of the cardiovascular system. Instead, there were twenty enzyme targets that could be hit to lower cholesterol production in the liver. The two most obvious enzymes to target were HMGR and SMO since they catalyze the two slowest steps in cholesterol anabolism. Through close to a century of experiments, effective inhibitors have been developed to lower LDL cholesterol and the incidence of both heart attacks and strokes.
While many successes have been made, there is still much work left to achieve the ambitious goal set by the WHO to reduce global cases of atherosclerotic cardiovascular diseases by 30% by 2030 [1]. This review detailed not only the overall pathway for cholesterol production but the structure and function of HMGR and SMO. We hope it led to an understanding of statin and terbinafine treatments. Great progress has been made in statin therapies, but additional work to improve SMO inhibition could provide alternative and perhaps more effective treatments of hypercholesterolemia. Such treatments would greatly improve the quality of life for millions of patients around the world.
Acknowledgements
This work was supported by a Departmental Research Grant from The Robert A. Welch Foundation AC-0006.
Footnotes
1Prokayotic HMGRs utilize NADH as reducing substrate while eukaryotes use NADPH. As is the case with many nicotinamide dependent enzyme cofactor specify does not alter the catalytic mechanism of the two classes of enzyme. The cofactor will be written as NAD (P) H as is customary in the field.
References
-
Ray KK, Ference BA, Tania S, Blom D, Nichollis S, et al. (2022) Worlds Health Federation Chlostero Roadmap 2022. Glob Heart 17(1): 75.
-
Watkins DA, Msemburi WT, Pickersgill SJ, Kawakatsu Y, Gheorghe A, et al. (2022) NCD Countdown 2030: efficient pathways and strategic investments to accelerate progress towards the Sustainable Development Goal target 3.4 in low-income and middle-income countries. Lancet 399(10331): 1266-1278.
-
Cory EJ, Russey WE, Montellano PRO (1966) 2,3-oxidosqualene, an intermediate in the biological synthesis of sterols from squalene. J Am Chem Soc 88(20): 4750-1751.
-
Feingold KR (2000) Introduction to Lipids and Lipoproteins. In: Feingold KR, et al. (Eds.), Endotext, South Dartmouth (MA), USA.
-
Smith LC, Pownall HJ, Gotto AM (1978) The plasma lipoproteins: structure and metabolism. Annu Rev Biochem 47: 751-757.
-
Hevonoja T, Pentikainen MO, Hyvonen MT, Kovanen PT, Korpela MA (2000) Structure of low density lipoprotein (LDL) particles: Basis for understanding molecular changes in modified LDL. Biochim Biophys Acta 1488(3): 189-210.
-
Bill CA, Vines CM (2020) Phospholipase C. Adv Exp Med Biol 1131: 215-242.
-
Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM (1994) The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem 45: 303-369.
-
Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A (2016) High Density Lipoproteins: Nature’s Multifunctional Nanoparticles. ACS Nano 10(3): 3015-3041.
-
Kaur N, Pandey A, Negi H, Shafiq N, Reddy S, et al. (2014) Effect of HDL-raising drugs on cardiovascular outcomes: a systematic review and meta-regression. PLoS One 9(4): e94585.
-
Keene D, Price C, Shin MJS, Francis DP (2014) Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117 411 patients. BMJ 349: g4379.
-
Bowman L, Hopewell J, Wiviott S, Sammons F, Chen F, et al. (2017) Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N Engl JMed 377(13): 1217-1227.
-
Singh P, Saxena R, Srinivas G, Pande G, Chattopadhyay A (2013) Cholesterol biosynthesis and homeostasis in regulation of the cell cycle. PLoS One 8(3): e58833.
-
Spady D, Woollett L, Dietschy J (1993) Regulation of plasma LDL-cholesterol levels by dietary cholesterol and fatty acids. Annual Rev Nutr 13(1): 355-381.
-
Grundy SM (1978) Cholesterol metabolism in man. West J Med 128(1): 13-25.
-
Bloch KE (1983) Sterol structure and membrane function. CRC Crit Rev Biochem 14(1): 47-92.
-
Kandutsch AA, Russell AE (1960) Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J Biol Chem 235: 2256-2261.
-
Pendse A, Lockyer P, Schisler J, Willis M (2011) Konrad Bloch, PhD: Understanding of Cholesterol Metabolism. Laboratory Medicine 42(6): 370-373.
-
Bloch K (1987) Summing up. Annu Rev Biochem 56: 1-19.
-
Langdon RG, Bloch K (1952) The Biosynthesis of Squalene and Chlosterol. J Am Chem Soc 74(7): 1869- 1870.
-
Miziorko HM (2011) Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys 505(2): 131-43.
-
Kritchevsky D, Moyer AW, Tesar WC, Logan JB, Brown RA, et al. (1954) Squalene Feeding in Experimental Atherosclerosis. Circulation Research 2(4): 340-343.
-
Nowosielski M, Hoffmann M, Wyrwicz LS, Stepniak P, Plewczynski DM, et al. (2011) Detailed Mechanism of Squalene Epoxidase Inhibition by Terbinafine. J Chem Info Model 51(2): 455-462.
-
Padyana AK, Gross S, Jin L, Cianchetta G, Narayanaswamy R, et al. (2019) Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nature Communications 10(1): 97.
-
Ornish D, Brown SE, Billings JH, Scherwitz LW, Armstrong WT, et al. (1990) Can lifestyle changes reverse coronary heart disease?: The Lifestyle Heart Trial. Lancet 336(8708): 129-133.
-
Brown MS, Goldstein JL (1986) A receptor-mediated pathway for cholesterol homeostasis. Science 232(4746): 34-47.
-
Gill JF, Beach MJ, Rodwell VW (1984) Transport of mevalonate by Pseudomonas sp. strain M. J Bacteriol 160(1): 294-298.
-
Haines BE, Wiest O, Stauffacher CV (2013) The increasingly complex mechanism of HMG-CoA reductase. Acc Chem Res 46(11): 2416-2426.
-
Lawrence CM, Rodwell VW, Stauffacher CV (1995) Crystal structure of Pseudomonas mevalonii HMG- CoA reductase at 3.0 angstrom resolution. Science 268(5218): 1758-1762.
-
Vogeli B, Shima S, Erb TJ, Wagner T (2019) Crystal structure of archaeal HMG-CoA reductase: insights into structural changes of the C-terminal helix of the class-I enzyme. FEBS Lett 593(5): 543-553.
-
Istvan ES, Palnitkar M, Buchanan SK, Deisenhofer J (2000) Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis. EMBO J 19(5): 819-830.
-
Tabernero L, Bochar DA, Rodwell VW, Stauffacher CV (1999) Substrate-induced closure of the flap domain in the ternary complex structures provides insights into the mechanism of catalysis by 3-hydroxy-3-methylglutaryl- CoA reductase. Proc Natl Acad Sci U S A 96(13): 7167- 7171.
-
Haines BE, Steussy CN, Stauffacher CV, Wiest O (2012) Molecular modeling of the reaction pathway and hydride transfer reactions of HMG-CoA reductase. Biochemistry 51(40): 7983-7995.
-
Steussy CN, Critchelow CJ, Schmidt T, Min JK, Wrensford LV, et al. (2013) A novel role for coenzyme A during hydride transfer in 3-hydroxy-3-methylglutaryl- coenzyme A reductase. Biochemistry 52(31): 5195- 5205.
-
Ono T, Bloch K (1975) Solubilization and partial characterization of rat liver squalene epoxidase. J Biol Chem 250(4): 1571-1579.
-
Berkel WJH, Kamerbeek NM, Fraaije MW (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol 124(4): 670-689.
-
Huijbers MME, Montersino S, Westphal AH, Tischler D, Berkel WJH (2014) Flavin dependent monooxygenases. Arch Biochem Biophys 544: 2-17.
-
Favre B, Ryder NS (1996) Characterization of squalene epoxidase activity from the dermatophyte Trichophyton rubrum and its inhibition by terbinafine and other antimycotic agents. Antimicrob Agents Chemother 40(2): 443-447.
-
Sakakibara J, Watanabe R, Kanai Y, Ono T (1995) Molecular Cloning and Expression of Rat Squalene Epoxidase. Journal of Biological Chemistry 270(1): 17- 20.
-
Abe I, Abe T, Lou W, Masuoka T, Noguchi H (2007) Site- directed mutagenesis of conserved aromatic residues in rat squalene epoxidase. Biochem Biophys Res Commun 352(1): 259-263.
-
Kantz A, Chin F, Nallamothu N, Nguyen T, Gassner GT (2005) Mechanism of flavin transfer and oxygen activation by the two-component flavoenzyme styrene monooxygenase. Arch Biochem Biophys 442(1): 102- 116.
-
Kantz A, Gassner GT (2011) Nature of the Reaction Intermediates in the Flavin Adenine Dinucleotide- Dependent Epoxidation Mechanism of Styrene Monooxygenase. Biochemistry 50(4): 523-532.
-
Buch K, Stransky H, Hager A (1995) FAD is a further essential cofactor of the NAD(P)H and O2-dependent zeaxanthin-epoxidase. FEBS Lett 376(1): 45-48.
-
Filippova TV, Blyumberg EA (1982) Mechanism of the Epoxidation of Alkenes by Molecular Oxygen. Russian Chemical Reviews 51(6): 582.
-
Brown MS, Danav SE, Siperstein MD (1974) Properties of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase Solubilized from Rat Liver and Hepatoma. J Biol Chem 249(20): 6585-6589.
-
Dietschy JM, Wilson JD (1970) Regulation of cholesterol metabolism. N Engl J Med 282(20): 1128-1138.
-
Endo A, Tsujita Y, Kursoda M, Tanzawa K (1977) Inhibition of Cholesterol Synthesis in vitro and in vivo by ML-236A and ML-236B, Competitive Inhibitors of 3-Hydroxy-3-methylglutaryl-Coenzyme A Reductase. Eur J Biochem 77(1): 31-36.
-
Yamamoto A, Sudo H, Endo A (1980) Therapeutic effects of ML-236B in primary hypercholesterolemia. Atherosclerosis 35(3): 259-266.
-
Endo A (2010) A historical perspective on the discovery of statins. Proc Jpn Acad Ser B Phys Biol Sci 86(5): 484- 93.
-
Alberts AW, Chen J, Kuron G, Hunt V, Huff J, et al. (1980) Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A 77(7): 3957-3961.
-
Endo A, Monacolin K (1979) A new hypocholesterolemic agent produced by a Monascus species. J Antibiot (Tokyo) 32(8): 852-854.
-
Tabernero L, Rodwell VW, Stauffacher CV (2003) Crystal structure of a statin bound to a class II hydroxymethylglutaryl-CoA reductase. J Biol Chem 278(22): 19933-19938.
-
Collins R, Reith C, Emberson J, Armitage J, Baigent C, et al. (2016) Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 388(10059): 2532- 2561.
-
Ruscica M, Ferri N, Banach M, Sirtori CR, Corsini A (2022) Side effects of statins: from pathophysiology and epidemiology to diagnostic and therapeutic implications. Cardiovasc Res 118(17): 3288-3304.
-
Alsuwaidan S, Alajlan A, Alkreathy H (2023) The effects of terbinafine on the lipid profile in humans and rabbits. Medical Science 27: 1-11.
-
Belter A, Skupinska M, Pietraszuk MG, Grabarkiewicz T, Rychlewski L, et al. (2011) Squalene monooxygenase - a target for hypercholesterolemic therapy. Biol Chem 392(12): 1053-1075.
-
Madan B, Virshup DM, Nes WD, Leaver DJ (2022) Unearthing the Janus-face cholesterogenesis pathways in cancer. Biochem Pharmacol 196: 114611.
-
Sagatova AA (2021) Strategies to Better Target Fungal Squalene Monooxygenase. J Fungi (Basel) 7(1): 49.
- Superposition of Cryo-EM and AlphaFold Predictions of Dengue Antigen-Antibody Complexes
- Jugular-Applied Coherent Low-Level Laser Therapy Enhances Systemic Mitochondrial Metabolic Function and Antioxidant Response
- Role of OMC32 Polypeptide in Acrosin-Mediated Exocytosis during the Bovine Sperm Acrosome Reaction
- Association of Galectin-3 but not Laminin in Tamoxifen-Induced Growth Suppression in Breast Cancer MCF-7 Cells
- Effect of Different Wavelengths of Light on the Rate of Photosynthesis
- Nutritional, Therapeutic, and Environmental Effect of Oyster Mushrooms: An Editorial