The Development of Tumor Heterogeneity and Actively Targeting Therapy Resistant Subpopulations
Tumor heterogeneity refers to the coexistence of cellular subpopulations bearing different epigenetic or genetic alterations within the tumor as a whole (intracellular tumor heterogeneity) and in different cells of the same patient referred to as secondary tumors/metastatic sites. This is clinically relevant because the bulk tumor mass may comprised of collection of cancer cells with varying levels of sensitivity to treatment and distinct molecular signatures. Tumor heterogeneity can also exist between the same cancer from different patients (inter-tumor heterogeneity) and within a single tumor (intra-tumor heterogeneity). Cancer cell heterogeneity introduces emerging challenges in classifying patients that might benefit from specific therapies or using molecular prognostic markers. Thus efforts in research for characterizing heterogeneity would be useful for a better understanding of the causes and progression of the disease. In the field of molecular diagnostics constituting tumor genotyping and blood based methods, recent advances allows the detection of clonal evolution in patients suffering from cancer.
Introduction
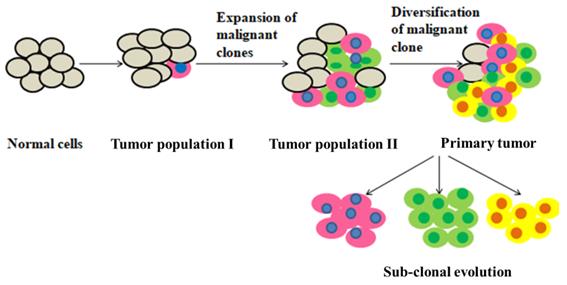
Tumor heterogeneity, namely the fundamental concept in which the architectural and molecular variability of tumor contributes to malignant phenotype poses a major challenge in clinical oncology [1]. Briefly, heterogeneity can exist between cancers from different patients (inter-tumoral heterogeneity) and within a single tumor (intra-tumor heterogeneity) [2]. Thus, inter- tumoral heterogeneity allows classifying tumor on the basis of molecular profiling, morphological characteristics and cell surface marker expression with respect to the same organ and/or between patients. Conversely, intra- tumor heterogeneity refers to variation within individual tumor and the unique tumor microenvironment (TME), which poses a considerable challenge in cancer therapy [3, 4]. Intra-tumoral heterogeneity and the unique TME characteristic of each patient contribute to the heterogeneity that makes each tumor unique as a result of the generation cellular clones. This causes the acquisition of resistance to several cancer therapies [4]. This in particular is a challenge in the advancement of precision medicine, which relies on a deeper understanding of cancer genes and mutational burden. With the advent of targeted therapies, tumor heterogeneity becomes increasingly interesting as this class of therapies is effective only in targeting events present in all cancer cells. Most drug development programs employing next- generation sequencing as a stratification tool do not consider the clonal or subclonal frequencies of a driver alteration, simply their presence or absence. Indeed, major targeted therapy strategies are in progress targeting the PI3K signaling axis despite evidence that somatic mutations in members of this pathway, including PTEN, PIK3CA, and mTOR, are often or always subclonal in ccRCC, ovarian, and prostate cancers. The clinical impact of driver variant allele frequency and the relative dominance of subclones with actionable alterations are priority areas for development within the context of clinical trial design. Emerging patterns of the temporal acquisition of mutations should further inform targeted therapy approaches. Until we have a greater understanding of complex paracrine and non-cell- autonomous interactions of cancer subclones, targeting a clonally dominant, truncal driver may provide a more effective drug development strategy than simply considering actionable alterations as present or absent. The DARWIN trial (Deciphering Anti-tumor Response with intratumor Heterogeneity (NCT02183883) aims to assess whether targeting a clonally dominant driver event results in improved progression free survival outcomes relative to targeting the same driver event when it is present subclonally. In addition, these studies will monitor the subclonal dynamics through therapy and during the acquisition of drug resistance [5]. Thus, it is important that novel drug development incorporates clonal heterogeneity to effectively target the complexity of tumors. In this chapter we outline the key aspects of tumor heterogeneity and current therapeutic options available for targeting these resistant sub-populations.
Tumor Heterogeneity
Intra-Tumor Heterogeneity
Intra-tumoral heterogeneity refers to the presence of distinct cancer cell subpopulations within tumors affecting malignant cells, additional cells within the tumor microenvironment (TME) and the extracellular matrix (ECM) [6, 7]. As a result, intra-tumoral heterogeneity can manifest as spatial heterogeneity due to an uneven distribution of genetically diverse tumor subpopulations and as temporal heterogeneity for the dynamic variations in genetic diversity of a particular individual tumor over time either as a consequence of natural tumor progression or as a consequence of exposure to selective pressures created by clinical interventions [8]. Exclusively, the mutations occurring in genes which are responsible for cell-cycle regulation and replication can result in genomic instability. For example, using temozolomide for the treatment in glioblastomas which can enhance transition mutations in mismatch repair (MMR) genes, results in the generation of hypermutated phenotype. Targeted therapy treatment might exert more potent selective pressures on oncogenic driven cancer cells compare to the therapies which are non-specific such as cytotoxic chemotherapy. Indeed, many of the most compelling observations regarding temporal heterogeneity have occurred within the context of treatment with targeted agents. Genetic and epigenetic variations in tumor cells and/or influence TME contributing to the development of intra-tumor heterogeneity [9]. Two models have been proposed to describe intra-tumor heterogeneity: clonal evolution model and cancer stem cell (CSC) model [10]. In clonal evolution model, all the undifferentiated clonal cells are thought to have similar tumorigenic ability, with mutations contributing to the adaptation of cells. This model explains intra-tumor heterogeneity as a result of natural selection, with the clones that acquire a growth advantage will survive and those that are less capable of surviving in a specific environment will be depleted. Over the course of disease, the clones may continue to change spatially and temporally, resulting in an even more complex sub-clonal architecture that is exacerbated by therapy [11]. Alternatively, the CSC model proposed that only a subset of cancer cells, that is CSCs, possess self- renewal capabilities and high proliferative potential to initiate and maintain tumor growth. This model suggests that CSCs contribute to heterogeneity in a hierarchical fashion comparable to normal tissue hierarchy by healthy stem cells. Although these clonal evolution model and the CSC model have been proposed as mutually exclusive explanations of intra-tumor heterogeneity, Peter C, et al. combined them in 1976, proposing a new model for cancer development. In this model, genomic instability of malignant cells resulted in the emergence of a multiclonal disease [2] Genetic events are not solely responsible for intra-tumor heterogeneity as characterization of the genetic profiles of several cancer cell lines revealed that the majority of the cell lines demonstrated a high degree of genetic homogeneity with drug response strongly being linked to intercellular epigenetic heterogeneity [12]. Epigenetic changes such as post-translational modification of histones, DNA methylation, and chromatin remodelling are important for the maintenance of genomic organization, normal cell function and gene expression [13]. Malignant cells exploit this intrinsic machinery in the development of resistance to therapy. Therefore, it is not surprising that epigenetic heterogeneity lead to an unsuccessful therapeutic response rate in cancer therapies [8] (Figure1).
Intrinsic Factors Affecting Intra-Tumoral Heterogeneity
Genomic instability is the most common and widely studied factor contributing to intra-tumoral heterogeneity. The Cancer Genome Atlas (TCGA) states that normal cells have a mutational rate of 10-9 mutations per base pair division, whereas the somatic mutation rate in cancer cells was 0.28 to 8.15 mutations per mega base suggesting high genomic instability in cancers [14]. Genomic alterations, which occur in the pathways of base excision repair, DNA mismatch repair, nucleotide excision repair, telomere maintenance, double-strand break repair, chromosome segregation and DNA replication, lead to extensive and stochastic changes across the entire genome [15]. Large scale genomic sequencing analyses have led to the identification of characteristic genetic signatures associated with some of these mutagenic processes [5, 16, 17]. In addition to genomic instability, epigenetic changes also play a major role in intra-tumor heterogeneity with epigenetic variations existing between tumor subpopulations. Several studies on cancer stem cells (CSCs) suggest that in a tumor, epigenetic changes occur in a limited number of cells in a fashion similar to normal stem cells and that differentiation of normal stem cells gives rise to non-tumorigenic cells which are phenotypically diverse having bulk of cells in a tumor. Although there is still no consensus regarding whether cancers arise from normal stem cells or whether there is a strict hierarchy or bidirectional plasticity between the differentiation and de-differentiation states. Most cancer biologists accept that at any given time in any type of cancer, there are populations of cells with self-renewal and tumor-initiating properties, which are simplified as stem-like cancer cells [15]. Another cause of intra-tumor heterogeneity is differential gene expression. In several studies plastic switching of the cellular state has been demonstrated [18, 19]. Specifically with the case of cancer cells, several cell surface antigens depending upon the drug treatment and tumorigenic assays undergo changes in a reversible manner, suggesting the mechanism of transient change in the gene expression [15]. Indeed, single-cell resolution experiments have demonstrated the stochastic gene expression as a fundamental nature of cells to cope with environmental changes, a common phenomena that is conserved from bacteria and phage till mammalian cells as well as cancer cells [20, 21]. Therefore, it is important to integrate all experimental approaches to fully understand the alterations at all levels and their distinctive roles in tumor progression.
Extrinsic Factors Affecting Intra-Tumor Heterogeneity
The extrinsic factors responsible for intra-tumor heterogeneity include the components of the surrounding microenvironment and originate from spatial differences in cancer cells. A tumors blood supply is an important factor that contribute to the architectural variations observed in the microenvironment [22]. Normally, nutrients, growth factors, and oxygen are delivered to cells and metabolic waste is removed through the blood vessels. Unlike in normal cells, the vascular network of tumor vessels is highly disordered, dilated and heterogeneously distributed [23, 24]. Intravital microscopy has revealed aberrant tumor vasculature as these blood vessels supply little or no oxygen [15]. The tumor core is generally hypovascular resulting in a chronically acidic and hypoxic region. The normal vascular network has a well-organized architecture, which provides nutrients by diffusion to all normal cells. In contrast, tumor blood vessels are dilated, saccular, tortuous and heterogeneously distributed. These structural and functional abnormalities in the tumor vasculature lead to a variable TME which in turn can lead to genomic instability, heterogeneous gene expression, signal transduction due to either indirectly via hypoxia, oxidative stress or acidosis or directly via hormones or growth factors [25]. Additionally, this microenvironment also creates an exclusive niche specifically enrich for CSCs in brain tumor [15]. In particular, it has been reported that the vasculature can provide a specialized growth niche for stem-like cancer cells in brain tumor. It has been reported that CD133+/Nestin+ cells of GBMs, medulloblastomas, ependymomas and oligodendrogliomas were located in close proximity to tumor capillaries. The self-renewal and proliferation properties of these cancer stem like cells were maintained in culture using factors secreted by endothelial cells [26]. Additional factors secreted by endothelial cells in culture also act to maintain the self-renewal and proliferative properties of CSCs [26]. Other components stromal cells such as inflammatory cells, mesenchymal, pluripotent cells, fibroblasts and components of the ECM secrete growth factors, cytokines which also contribute to the variability of genotypes and phenotypes [27].
Intertumoral Heterogeneity
Inter-tumoral heterogeneity, the cellular and molecular variations observed between tumors of the same origin, displaying different biological behaviours and unique molecular signatures that have variable impacts on clinical outcomes is driven by a combination of intrinsic and extrinsic mechanisms [8]. Intrinsic mechanisms of inter-tumoral heterogeneity include the genetic and epigenetic mutational profile of distinct cells within the tissue that leads to phenotypic differences. Additionally, extrinsic factors by the TME generate variability through complex interactions between the tumor and the TME, including immune cell infiltration, stromal heterogeneity or deregulation of the ECM [28].
Circulating Tumor Cells and Their Implication in Tumour Heterogeneity
Several reports suggests the mutational status differences of DNA and RNA or the expression profiles between the primary tumor and metastases, or within the single tumor [29, 30, 31, 32, 33]. Circulating tumor cells (CTCs) comprises a heterogeneous population of cells derived from tumor some of which may be the preliminary precursors of metastases or/and could result in spread of the primary tumor-to-metastasis or/and metastasis-to- metastasis. The CTCs which have been derived from the primary tumor or metastatic foci could invade the surrounding tissue, or enter the bloodstream or lymphatics, survives in circulation, extravasate into a different tissue and at last grow at the new site [34]. Several reports have provided robust evidence that CTCs represent the cellular heterogeneity determined by the spectrum of mutation in primary tumor and metastatic lesions much better than a localized primary tumor or metastatic biopsy [35, 36, 37]. Cell to cell diversity within CTCs have been shown in various studies [1, 38, 39]. Furthermore, the profiles of CTCs evolve gradually with the progression of disease [40, 41, 42]. Conventional molecular assays only reflect the signal from the dominant clone or an average signal from all the clones, even though this may not be the most malignant CTC clone. For this reason, single cell analysis may be a solution to this problem. Since the presence of CTCs is in critically lower concentrations in leukocytes in the periphery, the molecular characterisation of CTCs is challenging in the bloodstream. Various techniques such as DEParray™ technology (Silicon Biosystems, Bologna, Italy), the only automated instrument which identifies, quantifies and recovers individual cells and CellSearch® system (Veridex), which is the single assay for enumerating and enriching CTCs in clinics approved by FDA are used for the characterisation of CTCs [43, 44]. CTCs studies have focused on their prognostic significance, their utility in real-time therapies monitoring, mechanism of resistance, the identification of therapeutic targets, and understanding the process of metastasis [45]. It is shown that CTCs molecular characterisation is important for increasing the specificity for diagnosis and their potential as therapeutic targets. Recently, it has been shown that molecular characterisation of CTCs is pivotal for increasing the diagnostic specificity of CTCs and their potential as therapeutic targets [46]. Several studies have demonstrated CTCs are heterogeneous [47, 48, 49, 50]. Thus, the most reliable method established is the study of single- cell analysis of CTCs. Performing serial measurements of the dynamics of longitudinal biomarkers displayed in CTCs over the course of multiple sequential therapies, may provide insight into tumor evolution [51]. Also, temporal CTC monitoring may facilitate in identification of the effective drugs in individual cancer patients [52]; especially with those cases who already have tumor or will soon have tumor that are resistant to anti-cancer treatments. PIK3CA which is the most studied marker is one of the major examples. Studies reveal that in breast cancer, somatic mutations in PIK3CA play a major role in therapy response in breast cancer. Monitoring the mutational status of PIK3CA marker may thus improve the success of treatment decisions in these patients [53, 54].
The Development of Tumor Heterogeneity in Various Cancers
Breast Cancer Heterogeneity: Breast cancer is one of the leading causes of cancer death and most frequently diagnosed cancer in women worldwide [55]. It is one of the most heterogeneous forms of cancer and contributes to the challenges encountered in the clinic during prognosis where the complexity of this malignancy can be misjudged by a single biopsy. As such, pathologists take biopsies of multiple regions of a tumor and provide a diagnosis depending upon the region which is most malignant. However, despite this process, there is a likelihood of other aggressive regions not being overlooked due to their scarcity and/or topological heterogeneity [56]. Intra-tumor and inter-tumoral heterogeneity are characteristic which can complicate diagnosis and lead to challenges with elucidating an effective treatment regimen. The progression of breast cancer occurs from a premalignant disease (example; ductal carcinoma in situ (DCIS) or hyperplasia) to invasive carcinoma and metastasis. Histological classifications and molecular profiling provides an insight into the type or subtype of breast cancer that has developed in order to accurately treat a patient. Currently, the histological classifications of breast cancer include the presence or absence of progesterone-receptor, estrogen- receptor, and/ or ERBB2/HER2 whereas molecular profiling further subdivides breast cancer into at least six subtypes (normal, HER2 en-riched, claudin low, luminal A, luminal B and basal like) [57]. The different clinical prognosis is reflected by these expression profiles. Various concepts are proposed for the origin of diversity in the tumor such as cell plasticity and hierarchy in tumor cell, differentiation state of cell-of-origin, clonal cooperation, genetic evolution and tumor stroma as a source of heterogeneity in breast tumor [57]. The robustness of tumor is enhanced by heterogeneity which confuses diagnosis and prognosis and challenge cancer therapies [58, 59]. The dependency of heterogeneous breast tumor cells progression on a single pathway is rare and this is the basic cause of resistance to targeted therapies [57, 60]. A mutation which leads to resistance in the primary tumor may be present already at a very low frequency or may develop during the course of treatment. Despite the elimination of clones which were responsive, the clones which have become resistant may reduce the success of the treatment and result in tumor relapse and therapy failure [57]. Prostate cancer: Prostate cancer is the leading tumor of the male in Western societies [61]. The complicated diagnosis, therapy and prognosis of this tumour is difficult to the fact of heterogeneity in prostate cancer. The heterogeneous clones show different Resistance and sensibilities in various types of treatment. Transrectal sound examinations, digital rectal examination, determination of serum prostate specific antigen (PSA) are the common methods of identification and control of progression under treatment. However, these methods cannot predict further progression of the tumor at the beginning of the treatment because of the heterogeneity in prostate cancer. Certain tumors are androgen- independent and other are androgen-sensitive. DNA cytometry can be one of the mean to provide information about the biology of tumor and behaviour in prostate cancer [61]. Pancreatic cancer: Pancreatic adenocarcinoma is one of the deadliest cancers worldwide, especially due to its strong chemo-resistance and frequent diagnosis at an advanced stage. Tumor heterogeneity is significant at the histological level, within the tumor as well as between tumor. The most common of it is pancreatic ductal adenocarcinoma (PDAC). Besides that, epithelial tumor such as neuroendocrine tumor and acinar cell carcinoma also exist which have variable clinical outcomes and different molecular profiles, therefore should not be associated with PDAC. The prognosis of PDAC is very poor, with a 5-year survival rate of around 7%. Hence, a better and more effective molecular characterization approach for PDAC is necessarily required to identify the various pathways and key drivers resulting in carcinogenesis of pancreas and potential therapeutic targets. Various omic analysis such as metabolomics, transcriptomics, genomics have shown that the degree of PDAC heterogeneity is very high in terms of intra and inter-tumoral heterogeneity. PDAC identification with a deficiency in double strand break repair in DNA is routinely practiced and very challenging. The most appropriate technique will be targeted DNA sequencing approach with formalin-fixed paraffin-embedded samples. Another important method will be whole genome sequencing but it faces a lot of problem due to its expense and has a requirement of several bioinformatics structures for the analysis. However, reports suggests that similar approach in breast cancer could be effective on formalin-fixed paraffin-embedded biopsies with a higher sensitivity [62, 63]. Colorectal cancer: Colorectal cancer (CRC) is the second most common cancer in women and the third most common cancer in men. CRC varies at different levels leading to the differences in prognosis and therapeutic response, even with the cancers of same TNM stage. Because of these differences, strict system of classification is exploited in order to stratify colorectal tumor into various groups with direct prognostic and therapeutic response. Tumor heterogeneity has been studied at various levels such as histopathological features, transcriptomics, genomics, and characterization of inflammatory infilterate. KRAS, NRAS, BRAF mutation status is also investigated to determine the response of anti-EGFR therapy. Off lately, four significant molecular subtypes of colorectal cancer have been demonstrated having therapy and prognostic relevance. Considering together these parameters facilitate to stratify the patients into various prognostic and therapeutic subgroups [64]. Head and neck cancer: The majority of head and neck squamous cell carcinomas (HNSCC) arises from the upper aerodigestive tract at their mucosal linings, suggests a homogeneous disease. But, HNSCC are heterogeneous. Various bifurcations in terms of location, anatomic, etiology and molecular findings are present. The recent omics technology and various molecular biological approaches have led to the identification of HPV related HNSCC and resulted in the development of new classification of tumor. Better response rates to the established treatment modalities have been shown by HPV related oropharyngeal cancers shown leading to a better overall survival rate. Thus, the status of HPV is regarded as important prognostic biomarker in HNSCC, and immunostaining of p16 has become an important marker for the determination of the status of HPV. Despite of various improvements in the therapy and diagnosis, mortality and morbidity stayed high and desired treatment of patients is a major challenge [65].
“Go or Grow” Hypothesis
Traditionally, the tumour progression was assumed to be mutation driven. However, many experiments have shown a common association between the migratory and proliferative behaviour of cancer cells. Especially, proliferation and cell motion are mutually exclusive processes. It has been shown that only during resting phase when cells do not move, cell proliferation occurs. This phenomenon is known as ‘Go or Grow’ mechanism or migration/proliferation dichotomy [66, 67]. Biological evidence suggests that proliferative and migratory processes share common signalling pathway mechanism, indicating some unique intracellular mechanism which regulates both migration/proliferation behaviour. Such a hypothesis talks about the fact that migration (“go” as cell diffusion) and cell proliferation (“grow”) are mutually exclusive phenomena, at some point tumor cells stop mitosis to migrate [66]. Hypoxia has been proven to be a biological evidence for the switch from proliferative to an invasive phenotype supporting ‘go or grow’ hypothesis. Many biological studies have shown the association of hypoxia to the tumors aggressive behaviour. In particular, hypoxia has been shown to downregulate the levels cadherins, resulting in adhesive cell disruption and promotion of metastatic and invasive behaviour [68] and reduction in the proliferative activity. The reliance of the proliferation/invasion switch on the levels of oxygen suggests that the network responsible for controlling migration and mitosis and may share common signalling pathways with the oxygen uptake network [69]. A clear effect of tumor heterogeneity represented by these two specific phenotype is the diauxie-like curve controlling the total density of cell. This happens when the sub- population of migratory cells keep on increasing while the proliferative cells grow to a value which is maximum. Such result suggests the slow growth of cancer immunoediting process, when the immune system eliminates the cells which are modified followed by equilibrium and escape when tumor cells overcome suppression of immune system and tumor growth restarts with the activation of the migratory process [70, 71]. The association between ‘go or grow’ hypothesis and heterogeneity has not been explored much. Alexandre Sarmento Queiroga et al. demonstrated a critical approach for describing kinetics of the cell growth which recovers the sigmoidal-like cell densities. The sigmoidal model for example Gompertz model has been widely studied and the sigmoidal behaviour is associated with the depletion of resources in the culture experiments. They proposed the inverse relationship of the division rate with the migration rate and death rates suggesting the greater potential for cells to start migrating or dying when resources are scarce. The heterogeneity model demonstrated by Alexandre Sarmento Queiroga et al. was represented depending upon two major phenotypes, proliferative and other being predominantly migratory. This facilitated and shed light on how the diverse phenotypes coexisting in tumor affects its growth. Also, it facilitates in the characterization of the variability of spatial distribution of the sub-populations of cells of two tumors of the same size and having a potential refinement of cancer staging [72]. Therefore, it is very important to explore such a hypothesis and raise the idea of whether there is a relationship between tumor heterogeneity, migration and proliferation. Are there tumor subpopulations dedicated to proliferation and others dedicated to migration? How is it that the relationship between heterogeneity, proliferation and migration can affect the development of a tumor?.
Tackling the Complexity of Tumor Heterogeneity to Overcome Therapy Resistance Heterogeneity
Since Intra-tumor heterogeneity is responsible for the unsuccessful rate of the therapeutic response in cancer therapies, it is important to develop an effective approach to tackle this problem. Boyang Zhao et al. exploited an optimization approach to tackle special and temporal heterogeneity. Their data incorporated quantitative effects of targeted therapeutics and chemotherapeutic drugs which are commonly used on murine Eμ-myc; p19Arf-/-lymphoma cell line subpopulations generated by knockdown using shRNA in the apoptotic and DNA damage pathways. Computational analysis based on their generated data showed the utilisation of optimal drug combinations for tumors in which the genetic heterogeneity is qualitatively characterized. Initial result reflected that heterogeneity in tumor can homogenize drug efficacy effectively. Therefore, the drugs that kill the most possible broadest range of subpopulations are the most effective combinations of drug. Due to the fact of detection limitation of diagnostic tools and complexity within a tumor existing in patients poses a problem of incomplete information and uncertainty on the underlying composition of tumor for each patient. Irrespective of that, based on their sensitivity analysis and statistical sampling they discovered that particular combinations of set of drugs were directly linked with robustness and greater average efficacy. This suggests that even without the complete information of a tumor’s composition, a de novo optimal combinations of drug depending upon the robustness and average efficacy can be applied [73]. John Carl Panetta et al. have explored a mathematical model of heterogenous tumor populations where the resistance of this population to a chemotherapeutic treatment is also stimulated and have provided a medium of how such a model could improve such treatments. The considered mathematical model is a linear system of two ordinary differential equations which describes the cancer growth along with the effects of chemotherapy. The model considers the major parameters such as induction rate, dose period and growth rate. Consideration of these parameters is critical because if certainly the clinical data does not allow determining them, then the model will facilitate to develop an effective combination of chemotherapeutic treatments [74]. Also, Robinson F et al. described a nonlinear mathematical model for the cancer immunosurveillance focused on tumor cells phenotypic heterogeneity, especially regarding the recruitment capacities via cytotoxic activities, birth rates, angiogenic factors and immunogenicities differences. The model describes some invivo phenomenon such as robustness, tumor dormancy, immunoselection over cancer immunoediting in the tumor microenvironment composition. Bifurcation analysis gave tumor attractors fixed-points, limit cycles and chaotic attractors, the latter emerging from period-doubling cascade displaying Feigenbaum’s universality. Finally, simulation studies of both escape and elimination tumor scenarios by means of a stochastic version of the model according to the Doob- Gillespie algo algorithm [75].
Improving Diagnostic Methods to Identify Heterogeneity
The emergence and development of novel therapeutic strategies and clinical trials to target heterogenous subpopulations, particularly those that are resistant has recently begun to be explored. This is primarily because assessing the intra-tumoral heterogeneity was difficult and thus therapies targeting this heterogeneity could not be developed. Currently, liquid biopsies of patient blood samples contain cell-free circulating tumor DNA, circulating tumor cells can be used to perform DNA sequencing provides an opportunity for the assessment and careful measurement of determining heterogeneity in tumor. This method has been shown to be highly specific and sensitive and it can detect both sub-clonal and clonal mutations in addition to identifying the development of resistance earlier than radiographic imaging. However, liquid biopsies have limitations concerning the ability to assess spatial heterogeneity as such, a combination of liquid biopsies along with careful selection of image diagnostics, tissue biopsies and biomarkers can be a more effective method to carefully assess heterogeneity in tumor [76].
Targeting Cell Signalling Pathways
With more accurate identification of heterogeneity, specific pathways can potentially be targeted as a result including those that are vital to the cancer cells. For instance, in the pre-clinical models of the breast cancer cell lines having PIK3CA mutations, treatment with a PI3K/mTOR inhibitor, a single agent BEZ235, resulted in an apoptotic response only in the presence of the expression of BIM. However, irrespective of the expression of BIM, paclitaxel was similarly effective [77]. Besides this, the expression of various other factors such as heregulin can be raised by the mutation in PIK3CA, resulting in the activation of oncogenic pathways independent of PI3K [78]. It has also been shown that monotherapy with mTOR inhibitors can lead to feedback activation of the AKT signalling pathway [79, 80]. It has also been demonstrated that there is a co-existence of PIK3CA and KRAS mutation in patients suffering from advanced cancers. The preclinical data along with the early clinical observations suggests that the tumor having combined mutations could respond to combinations of MEK as well as PI3K mutations [81]. Also, in advanced stages of melanomas, inhibition of BRAF is effective having BRAF V600 mutations; but, having the same mutation in advanced colorectal cancer, treatment with inhibitor of BRAF vemurafenib resulted in dismal outcomes [81, 82]. Subsequently, in colorectal cancers, the preclinical outcomes showed various multiple resistant pathways, such as activation of EGFR, activation of PI3K pathway and abnormal methylation [83, 84].
Targeting Induction and Modulation and Apoptotic
The majority of the anti-cancer therapies indirectly or directly take advantage and exploits “apoptotic process” (programmed cell death) and various other pathways associated with cell death. However, as tumorigenesis progresses the acquisition of genetic alterations that to reduce sensitivity to cell death and under stress conditions increases their survival thereby minimizing the effectiveness of the drugs that target cell death [85, 86]. The majority of emerging therapeutic approaches focus on “reactivating” cell death programs. This reactivation of apoptosis can be initiated by two
| TNF-related apoptosis-inducing ligand | ||
|---|---|---|
| , | ( | TRAIL-R2/DR5) and DRs in melanoma, |
Therapeutically Targeting Epigenetic Changes that Contribute to Heterogeneity
Tumor cell plasticity and cellular transformation causing tumor heterogeneity require changes occurring at the epigenetic level through histone modification and DNA methylation. Such modifications in tumor cells are responsible for silencing tumor suppressor genes at various stages including development of drug resistance or Epithelial-mesenchymal transition (EMT) [93]. As such histone deacytylation via histone deacetylase (HDAC) and DNA methylation via DNA methyltransferases (DNMT) are potential therapeutic targets to be administered as combination therapy [94]. There are currently several drugs that target epigenetic regulators that are approved for treatment. Decitabine (5-azacytidine and 5-aza-2- deoxy-cytidine) (NCT01876641), a DNA hypomethylating agent and inhibitor of DNA Methyltransferase 1 (DNMT1) is currently clinically approved for hematological malignancies treatment [95]. The first histone deacetylase inhibitors (HDACi), vorinostat was approved as third line therapy for CTL (cutaneous T-cell lymphoma). Also, combination of vorinostat with pembrolizumab, a checkpoint inhibitor a form of cancer immunotherapy focusing on targeting immune checkpoints that stimulate or inhibit its actions, which tumor cells can utilise to protect themselves from attacks by the immune system is tested in patients suffering from advanced NSCLC (NCT02638090). In addition to these various other HDACi such as panobinostat, etinostat are currently being investigated for the treatment of lymphoma, melanoma and mesothelioma [96, 97].
DNA Damage Response Inhibitors
DNA damage response (DDR) mechanism are known to have consequences on processes such as DNA repair or cell cycle arrest or apoptosis [98]. In cancer, functional inactivation of DDR pathways lead to the increase in mutational load and genomic instability making the cells resistant to treatment. Thus. the use of DDR inhibitors is an important therapeutic approach. Considering the major therapeutic targets for inhibitors of DDR, currently being tested in clinical trials phase 0-II, are phosphatidyilinositol-3-OH-kinase (PI3K) family kinases, ataxia teleangiectasia mutated (ATM) and RAD3-related (ATR) and their downstream DDR kinases CHK1 and WEE1 [94]. Additionally, the inhibitors of the enzyme responsible for base excision repair (BER) of single strand breaks (SSB) known as poly (ADP-ribose) polymerase (PARP) are evaluated clinically as mono- or as combination therapy [99], Although with some limitations by using the inhibitors of DDR such as toxicity and lipophilicity in proliferating normal cells, the fundamental concept of synthetic lethality depending on DDR pathways mutations in tumor cells can be exploited therapeutically. Due to the increased sensitivity of tumor cells with inactivated BRCA 1 and BRCA-2 tumor suppressor genes towards the inhibition of PARP [94], the inhibitors of PARP such as rucaparib, olaparib and niraparib received the approval of FDA for the treatment of refractory breast cancers and BRCA-mutant ovarian cancer. In a similar manner, because of the p53/RB- pathway mutations creates a therapeutic window for CHK1 and ATR inhibitors and raises the sensitivity to DNA damaging drugs. The effectiveness of ATM inhibitors in preclinical studies in prostate and brain cancers has been observed in combination with radiotherapy as well as chemotherapy with increased sensitivity of PTEN-mutant or of p53-mutant cells [100]. With the case of melanoma, pre-clinical study suggest that PARP and DDR inhibition is a promising strategy for combinatorial therapy [94]. Therefore, in cells, the enhanced effects of DDR inhibitors having synergistic effects and sensitizing mutations of combination of various checkpoint inhibitors warrant further clinical testing.
Modulating Redox Activity
The reactive oxygen species (ROS) are defined as the cellular metabolism by- products and include compounds such as superoxides, peroxides, singlet oxygen which can cause damage to the basic biomolecules such as proteins and DNA. In tumor cells, the generation of ROS initiates the process of tumorigenesis by inducing DNA damage leading to mutations. These mutations can eventually cause metastasis and EMT by remodelling of the cytoskeleton and extracellular matrix. This process takes place over time and does not happen immediately following the DNA damage [101]. Due to the response of the oxidative stress, it hampers with various transcriptional regulators, enzymes responsible for mitochondrial detox and various tumor suppressors such as nuclear factor erythroid 2 (NRF2), NFκB therapeutic strategies targeting these processes with small molecules, phytochemicals or neutraceuticals may hold promise for targeted tumor combination therapy and may reduce cytotoxicity of conventional therapies Dimethyl fumarate (DMF), a redox modulator and is used as an approved treatment for several autoimmune diseases such as psoriasis and multiple sclerosis, induces recycling of intracellular glutathione (GSH) pool, inhibits NFκB p65/RelA thereby enhancing the overall response of anti- oxidant properties in various cell types [102]. DMF has the potential to cause programmed cell death in cells with T cell lymphoma [103] which is currently under phase-1 clinical trial for refractory chronic lymphocytic leukemia (CLL) (NCT02784834). In melanoma, the pro-apoptotic effects and the anti-proliferative effects of DMF have shown to reduced metastasis and growth of melanoma in pre-clinical models [100]. Similiarly, thiadiazolidinone (TDZD-8), a GSK-3β inhibitor, has proven to be effective in causing death in leukemia cells exhibiting stem cell-like markers [104]. The proposed mechanism of this is thought to via eliminating thiols with quick accumulation of ROS- causing minimal toxicity to normal hematopoietic cells. An alternative anti-cancer therapy includes the use of vitamins having anti-oxidant properties and is under phase-II clinical trial evaluating the effectiveness of higher doses of sodium ascorbate (Vitamin C) along with chemotherapy to kill heterogenetic tumor cell populations and to reduce toxic side effects (NCT02655913) [105]. Besides these, various other anti- oxidants and vitamins such as catechins, vitamin E, N- acetylcysteine (NAC) are been evaluated along with the combination therapies [94].
Metabolic Drugs
Another approach to target heterogenous populations is interfering with metabolic reprogramming in cancer cells focussing on major pathways such as glycolysis and various energy metabolisms. These strategies involve anti-diabetic biguanides phenformin and metformin which have shown anti-cancer properties by interfering with inhibition of mitochondrial complex and mTOR signalling pathway [106]. The mitochondrial ATP production and intermediates of tricarbocylic acid (TCA) cycle are shown to be decreased in the presence of biguanides. This lead to cancer cell death by reducing available glycolysis produced leading to cancer cell death by reducing available glycolysis produced ATP, availability to tumor cells [107]. Additionally, a pyruvate mimetic compound dichloroacetate (DCA), a metabolic drug with anti-cancer properties is used as a treatment for pediatric mitochondrial disorders [108]. This lead to the stimulation of function of mitochondria by inhibiting PDK1-4 (pyruvate dehydrogenase kinases) at the expense of glycolysis to reverse Warburg effect and suppress the tumor cell growth advantage [108]. Clinical trials examining the effect of DCA vs. Placebo in combination with cisplatin and treatment in radiation in patients suffering from III-IV stage of Head and neck squamous cell carcinoma (HNSCC) are currently underway (NCT01386632) [94].
Targeting Cancer Evolution
Genomic Instability Modulation
Epigenetic and genetic factors lead to tumor evolution resulting in reduced genomic stability. Examples such as BRCA1 and BRCA2 deficient breast and ovarian cancers lacking repair in homologous recombination, mismatch repair proteins function loss in sporadic and familial colorectal cancers [109]. Recently, the APOBEC family involved in protein activation has been shown to increase the rate of mutation across half of cancers [110] in humans and represents a common cause of subclonal diversification in NSCLC [111]. However, alterations in genes responsible for controlling stability of the genome have not been tested. A well-known example to kill homologous recombination-deficient cells is to use synthetic lethal approach as shown by the inhibitors activity of PARP in BRCA- like and BRCA- deficient cells [108]. This reflects that increasing instability in the genome by targeting a complementary DNA repair pathway over the tolerability threshold might result in breakdown of integrity of the genomic and consequently to cell death [112]. Additionally, a very high response rate to the immune checkpoint inhibitors is observed with the tumors bearing mismatch deficiency, thus significantly matching the levels of mutational burden with respect to the therapeutic efficacy [113]. Analogously, controlling instability in the genome for therapeutic purposes may slow down the progression in the tumor [114].
Targeting Clonal Mutations
Since the heterogeneity of the subclones of tumor occur at the epigenetic or genetic level factors evolution under selective pressure of anticancer drugs, it would be intuitive to think that in the cells the administration of drugs targeting truncal alterations could better increase the odds of durable control of disease [112]. In this regard, Pearson, et al. have demonstrated that the patients suffering from gastric cancer harboured tumor with a high level of FGFR clonal amplifications when treated with FGFR inhibitor AZD4547. Conversely, subclonal or low level of amplification was harboured in tumor which did not respond [115]. Studies involving 120 patients suffering from breast cancer and undergoing treatment with PI3K/AKT/mTOR inhibitors, tumor having clonal mutations in PIK3CA gene expressed a trend towards a improved and better response, which was however not statistically significant (82); indeed, as reported in study based on TCGA by McGranahan and colleagues, the raised frequency of alterations at the subclonal level of PI3K/mTOR across various tumors type, reflects that this could at least partially account for the modest results seen with the inhibitors of PI3K in patients with solid malignancies. In a similar fashion, in individual cancers careful knowledge of the status of clones of actionable could benefit the design and implementation of various therapies preventing it from acquired resistance. However, it is not always feasible to target clonal alterations directly. This is with the case of loss of function of adenomatous polyposis coli (APC), a tumor suppressor. In preclinical models apoptosis occurs after the restoration of APC in colorectal cancer cell line and regression of tumor [116]. The restoration of APC activity at the pharmacological level has unfortunately yet to be investigated. However, p53 in various tumor types which is distinctly enhanced in clonal mutations, restoration of p53 lead to regression of tumor in autochthonous mouse sarcomas and lymphomas [112].
Immunotherapy
The interplay role between the host immune system and a tumor can facilitate in identification of the tumor immunogenicity [117]. The process of immunosurveillance can select for subclones with limited sensitivity to immune attack or subclone lacking immunogenic antigens [118] mediating evolution of clones and progression of tumor, a process termed as immunoediting [119]. Tumor cells can generate a microenvironment that actively captures the antitumor immune response of host by using various suppressive mechanisms such as the expression of indolamine 2,3- dioxygenase, programmed cell death ligand 1 (PDL1), the induction of Tcell anergy or the secretion of anti- inflammatory mediators (i.e., TGFb) [117]. Also, tumor have the potential to attract various other types of cells which would suppress invading immune cells and facilitate the growth of the tumor as well as survival, such as tumor-associated macrophages (TAM), cancer- associated fibroblasts (CAF), regulatory T cells, and myeloid derived suppressor cells (MDSC) [117]. Drugs which are immunotherapeutic such as cancer vaccines and monoclonal antibodies have become an important player in the landscape of cancer therapies and in the treatment of tumor heterogeneity. Also, as compared to the conventional therapies, immunotherapy plays a major important role in its potential to initiate immunological memory and epitope spreading, which may significantly contribute to preventing disease relapse. Especially, immunoregulatory checkpoints modulation has recently been illustrated in several clinical trials in which antibody-mediated blockade of the immune inhibitory receptors programmed cell death ligand-1 (PD-L1), programmed cell death protein-1 (PD-1), or cytotoxic T- lymphocyte–associated protein 4 (CTLA-4) resulted in significant benefits at the clinical level in various types of cancers [120–122]. TILs, particularly CD8+ T cells, have been shown to be in association with improved prognosis as well as treatment response various tumor types such as ovarian, melanoma, breast and lung. The mutant peptide immunogenicity relies on its binding affinity with MHC class I ligands for presentation and recognization by CD8+ T cells [117]. Toxicity and druggability is the major issue when targeting with agents affecting multiple clonal alterations. To overcome these limitations various strategies are involved such as targeting dominant branched antigens or clonal neoantigens which were determined via evolutionary strategies such as systemic therapy or surgery, through adoptive cell therapy or vaccines. Snyder, et al. identified in a study of patients with melanoma treated with CTLA-4 blockade (ipilimumab or tremelimumab), candidate tumor neo- antigens for each patient and validated a neo-antigen signature associated with a strong treatment response [117]. Such studies have the potential to use the neo- antigen landscape of heterogeneous tumor to predict immunotherapy response and therefore aid treatment stratification.
Regulating tumor metabolism for the improvement of immune suppression of tumor microenvironment is one of the widely studied areas of research. Inhibition of Indoleamine 2,3-dioxygenase (IDO) with immunotherapy has shown effective results. Currently, two primary drug types against IDO are exploited: i) IDO inhibitor which is highly potent and inhibits tryptophan degradation example epacadostat [123] and ii) the pathway inhibitor of IDO which reverses IDO-mediated suppression of immune system and inhibits tryptophan degradation [124]. Also, in recent clinical trials, the clinical efficacy and safety of these two types of drug have been confirmed [125, 126]. Additionally, the inhibitor of IDO; epacadostat in combination with pembrolizumab, anti-PD1 antibody have been shown as a safe and effective medium for advanced cancer types in clinical trials. Furthermore, clinical trial is going on for indoximod in combination with immune checkpoint inhibitor (NCT02073123) to study their preliminary efficacy. Therefore, it is predictable that IDO inhibitors have a potential synergistic effect with checkpoint inhibitors of immune system [127]. As a consequence of altered tumor metabolism, hypoxic and acidic microenvironments are caused within the tumor causing heterogeneity. Therefore, targeting hypoxic and acidic microenvironment is widely exploited as a strategy to modulate tumor microenvironment. It has been shown that PX-478, a selective inhibitor causes the suppression of Hypoxia inducible factor (HIF-1α) levels [128]. Also, upregulation of lactate levels can significantly upregulate Bcl-2 levels via the translational control mediating starvation of glucose in cancer cells and promoting resistance. Cisplatin and etoposide in the clinical trial during abnormal metabolism of lactate in combination with Bcl-2 inhibitor AT-101 enhanced the antitumor effect [129]. Additionally, Monocarboxylate transport inhibitor AZD3965 mediating the transport of lactate, pyruvate and other metabolites showed effective antitumor effect [130].
Immunosuppressive conditions within the tumor can be modulated by the regulation of heterogenous components in stroma. The major underlying mechanism to reduce proliferation and invasion of tumor includes weakening the function of cancer associated fibroblasts (CAF), normalization of blood vessels within tumor for the effective transportation of drugs or immune cells to the specific tumor sites [131]. Considerable efforts are being taken for the development of anti-angiogenic drugs and several drugs have already been approved by FDA such as Avastin (bevacizumab) [132]. The combination of IFN-α and bevacizumab has passed clinical trial phase 2 and 3 and have been demonstrated to improve clinical efficacy in metastatic renal cell carcinoma suggesting the importance of combinatorial therapy over monotherapy. Also, clinical trials are going on for apatinib, the VEGF receptor tyrosine kinase inhibitor and PD-1 inhibitor to illustrate the efficacy for the treatment of gastric cancer types. Several studies are also focussing on targeting CAF to improve immunosuppressive condition by modulating CAF in the tumor microenvironment responsible for causing heterogeneity. It has been shown that the humanized monoclonal antibody sibrotuzumab directed against FAPα on CAFs has the potential to inhibit dual function of protease and signal transduction and thereby could block tumor progression, invasion and metastasis.
Recently, combining traditional treatment methodologies such as radiotherapy and chemotherapy with immunotherapy is receiving global attention. Every chemotherapeutic drug has a different underlying immunological mechanism associated with it for better efficacy. For example; drugs such as, 5-fluorouracil (5-Fu), oxaliplatin and anthracycline resulted in tumor cell apoptosis thereby increasing immunogenicity. Drugs such as paclitaxel, gemcitabine and pemetrexed caused direct immunostimulation mediating tumor immunity of immune effector cells. Therefore, radiotherapy and chemotherapy can facilitate the elimination of number of tumor cells, and expose large number of neoantigens and tumor antigen products in the microenvironment which could recruit large number of immune effector cells and at last improve the immunosuppressive state of the tumor microenvironment [133].
Combinatorial Therapy Approach
The combinatorial approaches of drugs which are effective lately support the effectiveness of chemotherapeutic regimens. The approach of combined therapy is further supported by various models of acquired resistance; such as on patients with colorectal, melanoma, pancreatic cancer suggests that with appropriate targeted agents, monotherapy alone cannot abolish the disease in metastatic cancer even if there is limited tumor burden pressure. However, dual approach might be successful only with the case if the tumor burden is low and in absence of cross- resistance mutations. In Colorectal cancers, for example, secondary resistance to anti-EGFR antibody therapy, which interferes with signalling via the MAPK cascade, is often led by pathway reactivation through additional alteration via gain-of- function mechanism MEK, MET and RAS [134]. Following these observations, Misale, et al. showed that combinatorial treatment of EGFR-sensitive colorectal cancer models with vertical inhibition of EGFR and MEK, downstream effector of MAPK pathway prevents the occurrence of resistance [134] and a clinical trial adopting this approach in EGFR-sensitive CRC is ongoing to test the hypothesis (EudraCT 2014-00246033). Henceforth, combination of three or higher order combinatorial therapies is important for eradication of tumor even with the agents targeting truncal alterations; analogously, inhibition of distinct pathways would also be required to avoid cross-resistance [112].
Clinical Implications and Future Directions of Targeting Heterogeneity
The leading cause for treatment failure can be attributed to tumor heterogeneity. The variants of biomarkers in intra and inter-tumoral heterogeneity present challenge when using these biomarkers for patient stratification with the intention of predicting therapy response and shows spatial variability. Additionally, metastatic tumor cells and variable microenvironment at metastatic sites, divergent evolution may lead to change in the expression of the biomarker in the primary tumor which was initially identified [135]. Hence depending upon the expression of biomarkers, treating the metastatic disease in the primary tumor may thus be less than optimal. Better understanding of changes in populations of cells and monitoring content of the tumor during the progression of disease and treatment may improve the diagnosis of cancer and therapeutic design. During patient’s treatment, carefully visualising plastic or genetic changes in cancer might lead to the well selection of the next therapy as well as the subsequent therapy. To capture spatial heterogeneity; researchers would acquire multiple regions from each tumor or metastasis. Several other analyses including epigenetic are also important to be performed. Another necessary step is phenotypic correlation and clinical annotation of the samples [1, 45]. In recent times, it has been shown that it is very important for the molecular characterisation of CTCs for their potential therapeutic targets. Since it has been shown in various studies the heterogenous nature of CTCs [45], the most reliable method established is single-cell analysis of CTCs. Another method which may help to detect the effective drugs in individual cancer patients is monitoring temporal heterogeneity [53] of CTCs exclusively for those who is already suffering from tumor or have chances for the development of tumors which are resistant to anti- cancer treatments. A classic example is PIK3CA which is the most studied marker. In the response therapy in breast cancer somatic mutation in PIK3CA plays a very important role. Examining the mutational status of PIK3CA marker might improve the further triumph of the treatment regime in the patients suffering from cancer [54]. In summary, A variety of small-molecule targeted therapies are currently available for novel combinational and alternative therapy regimens based on individual patient and tumor parameters considering heterogeneity. Therefore, it is now important to establish additional biomarkers including gene mutation patterns, expression changes, epigenetic makers, liquid markers, and tumor micro-environmental markers-to improve the prediction of the best combination regimen for each patient, tumor, and therapeutic stage. In the treatment of advanced melanoma, both additional triple combinations with BRAFi and MEKi and successive application of targeted therapies, as well as combinations of small-molecules with immunotherapy, chemotherapy, and radiotherapy may open novel perspectives. Furthermore, identification of additional tumor biomarkers may also lead to improved prevention and adjuvant strategies [54].
References
-
Tellez Gabriel M, Ory B, Lamoureux F, Heymann MF, Heymann D (2016) Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis. Int J Mol Sci 17(12): 2142.
-
Prasetyanti PR, Medema JP (2017) Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer 16(1): 1-9.
-
Visvader JE (2011) Cells of origin in cancer. Nature 469(7330): 314-322.
-
Franziska M, Kornelia P (2010) The Origins and Implications of Intratumor Heterogeneity. Cancer Prev Res 3(11): 1361-1364.
-
Mcgranahan N, Swanton C (2015) Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell 27(1): 15-26.
-
Hayes DF, Paoletti C (2013) Circulating tumour cells : insights into tumour heterogeneity. J Intern Med 274(2): 137-143.
-
Friedl P, Alexander S (2011) Review Cancer Invasion and the Microenvironment : Plasticity and Reciprocity. Cell 147(5): 992-1009.
-
Dagogo jack I, Shaw AT (2018) Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 15(2): 81-94.
-
Nassar A, Radhakrishnan A, Cabrero IA, Cotsonis GA, Cohen C (2010) Intratumoral Heterogeneity of Immunohistochemical Marker Expression in Breast Carcinoma: A tissue microarray-based study. Appl Immunohistochem Mol Morphol 18(5): 433-441.
-
Visvader JE, Lindeman GJ (2008) Cancer stem cells in solid tumours : accumulating evidence and unresolved questions. Nat Rev Cancer 8(10): 755- 768.
-
Waclaw B, Bozic I, Pittman ME, Hruban RH (2015) A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 525(7568): 261-264.
-
Mazor T, Pankov A, Song JS, Costello JF (2016) Intratumoral heterogeneity of the epigenome. Cancer cell 29(4): 440-451.
-
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28(10): 1057- 1068.
-
Kandoth C, Mclellan MD, Vandin F, Ye K, Niu B, et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature 502(7471): 333-339.
-
Sun X, Yu Q (2015) Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Nat Publ Gr 36(10): 1219-1227.
-
Pleasance ED, Stephens PJ, Meara SO, Mcbride DJ, Meynert A, et al. (2010) A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 463(7278): 184-190.
-
Govindan R, Ding Li, Griffith M, Subramanian J, Dees ND, et al. (2012) Genomic landscape of non-small cell lung cancer in smokers and never smokers. Cell 150(6): 1121-1134.
-
Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, et al. (2011) Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 146(4): 633-644.
-
Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, et al. (2014) Dynamic Heterogeneity and DNA Methylation in Embryonic Stem Cells Mol Cell 55(2): 319-331.
-
Raj A, Oudenaarden AV (2011) Stochastic gene expression and its consequences. Cell 135(2): 216- 226.
-
Fukumura D, Duda DJ, Munn LL, Jain RK (2010) Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre- clinical models. Microcirculation 17(3): 206-225
-
Brown EB, Campbell RB, Tsuzuki Y, Carmeliet P (2001) In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat Med 7(7): 864-868.
-
Jain RK (1988) Review Determinants of Tumor Blood Flow : a Review. Cancer res 48(10): 2641-2658.
-
Gillies RJ, Verduzco D, Gatenby RA (2014) Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 12(7): 487-493.
-
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, et al. (2007) A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 11(1): 69-82.
-
Junttila MR, De Sauvage FJ (2013) Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501(7467): 346-354.
-
Cassidy JW, Caldas C, Bruna A (2015) Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer res 75(15): 2963-2968.
-
Vermaat JS, Nijman IJ, Koudijs MJ, Gerritse FL, Scherer SJ, et al. (2012) Primary Colorectal Cancers and Their Subsequent Hepatic Metastases Are Genetically Different : Implications for Selection of Patients for Targeted Treatment. Clin Cancer res 18(3): 688-700.
-
Thomas A, Rajan A, Giaccone G (2012) Tyrosine Kinase Inhibitors of Lung Cancer. Hematol Oncol Clin North Am 26(3): 589-605.
-
Mao C, Wu X, Yang Z, Threapleton DE, Yuan JQ, et al. (2015) Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci Rep 5: 8065.
-
Colombino M, Capone M, Lissia A, Cossu A, Rubino C, et al. (2019) BRAF / NRAS Mutation Frequencies Among Primary Tumors and Metastases in Patients With Melanoma. J Clin Oncol 30(20): 2522-2529.
-
Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, et al. (2012) Comparative Genomic Analysis of Primary Versus Metastatic Colorectal Carcinomas. J Clin Oncol 30(24): 2956-2562.
-
Steeg PS (2006) Tumor metastasis : mechanistic insights and clinical challenges. Nat Med 12(8): 895- 904.
-
Tellez Gabriel M, Heymann M, Heymann D (2019) Circulating Tumor Cells as a Tool for Assessing Tumor Heterogeneity. Theranostics 9(16): 4580- 4594.
-
Suhaimi NM, Miin Y, Yoke D, Lee S, Phyo WM, et al. (2015) Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol Oncol 9(4): 850-860.
-
Mostert B, Jiang Y, Sieuwerts AM, Wang H, Bolt-de Vries J, et al. (2013) KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int J Cancer 133(1): 130-141.
-
Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, et al. (2013) Complex Tumor Genomes Inferred from Single Circulating Tumor Cells by Array-CGH and Next-Generation Sequencing. Cancer Res 73(10): 2965-2975.
-
Solakoglu O, Maierhofer C, Lahr G, Breit E, , Scheunemann P, et al. (2002) Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc Natl Acad Sci USA 99(4): 2246-2251.
-
Pailler E, Auger N, Lindsay R, Veilh P, Islas-Morris- Hernandez A, et al. (2015) High level of chromosomal instability in circulating tumor cells of ROS1- rearranged non-small-cell lung cancer. Ann Oncol 26(7): 1408-1415.
-
Middleton G, Crack LR, Popat S, Swanton C, Hollingsworth SJ, et al. (2015) The National Lung Matrix Trial : translating the biology of stratification in advanced non-small-cell lung cancer. Ann Oncol 26(12): 2464-2469.
-
Jamal hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, et al. (2014) Tracking Genomic Cancer Evolution for Precision Medicine : The Lung TRACERx Study. PLoS Biol 12(7): e1001906.
-
Joosse SA, Gorges TM, Pantel K (2015) Biology, detection and clinical implications of circulating tumor cells. EMBO Mol Med 7(1): 1-11.
-
Gabriel MT, Calleja LR, Chalopin A, Ory B, Heymann D (2016) Circulating Tumor Cells : A Review of Non EpCAM-Based Approaches for Cell Enrichment and Isolation. Clin Chem 62(4): 571-581.
-
Alix panabie C, Pantel K (2013) Circulating Tumor Cells : Liquid Biopsy of Cancer. Clin Chem 59(1): 110- 118
-
Toss A, Mu Z, Fernandez S, Cristofanilli M (2014) CTC enumeration and characterization : moving toward personalized medicine. Ann Transl Med 2(11): 108.
-
Sakaizawa K, Goto Y, Kiniwa Y, Uchiyama A, Harada K, et al. (2012) Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br J Cancer 106(5): 939-946.
-
Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, et al. (2013) Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci USA 110(52): 21083-21088.
-
Fernandez SV, Bingham C, Fittipaldi P, Austin L, Palazzo J, et al. (2014) TP53 mutations detected in circulating tumor cells present in the blood of metastatic triple negative breast cancer patients. Breast Cancer Res 16(5): 445.
-
Pestrin M, Salvianti F, Galardi F, De Luca F, Turner N, et al. (2014) Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol Oncol 9(4): 749-757.
-
Nakazawa M, Lu C, Chen Y, Paller CJ, Carducci MA, et al. (2015) Serial blood-based analysis of AR-V7 in men with advanced prostate cancer. Ann Oncol 26(9): 1859-1865.
-
Hwang W, Hwang K, Miyamoto DT (2016) The promise of circulating tumor cells for precision cancer therapy. Biomark Med 10(12): 1269-1285.
-
Kobayash S, Canepa HM, Bailey AS, Yamaguchi N, Goldstein MA, et al. (2013) Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J Thorac Oncol 8(1): 45-51.
-
Markou A, Schiza C, Efstathiou T, Kounelis S, Malamos N, et al. (2014) PIK3CA Mutational Status in Circulating Tumor Cells Can Change During Disease Recurrence or Progression in Patients with Breast Cancer. Clin Cancer Res 20(22): 5823-5834.
-
Jamal hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, et al. (2014) Tracking Genomic Cancer Evolution for Precision Medicine: The Lung TRACERx Study. PLoS Biol 12(7): e1001906.
-
Komaki K, Sano N, Tangoku A (2006) Problems in Histological Grading of Malignancy and its Clinical Significance in Patients with Operable Breast Cancer. Breast Cancer 13(3): 249-253.
-
Koren S, Bentires-alj M (2015) Review Breast Tumor Heterogeneity : Source of Fitness, Hurdle for Therapy. Mol Cell 60(4): 537-546.
-
Zardavas D, Baselga J, Piccart M (2013) Emerging targeted agents in metastatic breast cancer. Nat Rev Clin Oncol 10(4): 191-210.
-
Diaz Cano SJ (2012) Tumor Heterogeneity: Mechanisms and Bases for a reliable Application of Molecular Marker design. Int J Mol Sci 13(2): 1951- 2011.
-
De Palma M, Hanahan D (2012) The biology of personalized cancer medicine : Facing individual complexities underlying hallmark capabilities. Mol Oncol. 6(2): 111-127
-
Landis SH, Murray T, Bolden S, Wingo PA (1999) Cancer Statistics 1999. CA Cancer J Clin 49(1): 8-31.
-
Krause FS, Feil G, Bichler K, Schrott KM, Akcetin ZY, et al. (2005) Heterogeneity in Prostate Cancer: Prostate Specific Antigen ( PSA ) and DNA Cytophotometry. Anticancer Res 1786 (2005): 1783-1785.
-
Cros J, Raffenne J, Couvelard A, Pote N (2018) Tumor Heterogeneity in pancreatic adenocarcinoma. Pathobiology 85(1-2): 64-71.
-
Sagaert X, Vanstapel A, Verbeek S (2018) Tumor Heterogeneity in Colorectal Cancer : What Do We Know So Far ?. Pathobiology 85(1-2): 72-84.
-
Klussmann JP (2017) Head and Neck Cancer New Insights into a Heterogeneous Disease. Oncol Res Treat 40(6): 318-319.
-
Hathzikirou H, Basanta D, Simon M, Schaller K, Deutsch A (2012) 'Go or grow': the key to the emergence of invasion in tumour progression?. Math Med Biol. 29(1): 49-65.
-
Giese A, Loo MA, Tra N, Haskett D, Coons SW, et al. (1996) Dichotomy of astrocytoma migration and proliferation. Int J Cancer 67(2): 275-282.
-
Sullivan R, Graham CH (2007) Hypoxia-driven selection of the metastatic phenotype. Cancer Metastasis rev 26(2): 319-331.
-
Eckerich C, Zapf S, Fillbrandt R, Loges S, Westphal M, et al. (2007) Hypoxia can induce c-Met expression in glioma cells and enhance SF / HGF-induced cell migration. Int J Cancer 121(2): 276-283.
-
Reilly EO, Tirincsi A, Logue SE, Szegezdi E (2016) The Janus Face of Death Receptor Signaling during Tumor immunoediting. Front Immunol 7: 446.
-
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting : from immuno- surveillance to tumor escape. Nat Immunol 3(11): 991-998.
-
Citrangulo T, Sarmento QA, Chammas A (2019) A stochastic spatial model for heterogeneity in cancer growth. bioRxiv pp: 1-14.
-
Zhao B, Hemann MT, Lauffenburger DA (2014) Intratumor heterogeneity alters most effective drugs in designed combinations. Proc Natl Acad Sci USA 111(29): 10773-10778.
-
Panetta JC (1998) A Mathematical Model of Drug Resistance : Heterogeneous Tumors. Math Biosci 147(1): 41-61.
-
Alvarez RF, Barbuto AM, Venegeroles R (2019) A nonlinear mathematical model of cell-mediated immune response for tumor phenotypic heterogeneity. J Theor Biol 471(21): 42-50.
-
De Lartigue J (2018) Tumor heterogeneity: a central foe in the war of cancer. 16(3): e167-e174.
-
Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, et al. (2014) mTOR Inhibition Specifically Sensitizes Colorectal Cancers with KRAS or BRAF Mutations to BCL-2/BCL-XL Inhibition by Suppressing MCL-1. Cancer Discov 4(1): 42-52.
-
Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, et al. (2011) H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene 29(37): 5193-5203.
-
Carracedo A, Ma L, Teruya-feldstein J, Rojo F, Salmena L, et al. (2008) Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 118(9): 3065-3074.
-
Rodrik Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, et al. (2011) mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov 1(3): 248- 259.
-
Janku F (2018) Tumor heterogeneity in the clinic : is it a real problem ? Ther Adv Med Oncol 6(2): 43-51.
-
Mcarthur A, Flaherty KT, Puzanov I, Ribas A, Chapman PB, et al. (2013) Inhibition of Mutate Activated BRAF in Metastatic Melanoma. N Engl J Med 363(9): 809- 819.
-
Dayyani F, Nanda VGY, Jiang Z, Wistuba II, Xi M (2013) Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res 19(3): 657- 667.
-
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, et al. (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483(7387): 100-103.
-
Fouad YA, Aanei C (2017) Revisiting the hallmarks of cancer. Am J Cancer Res 7(5): 1016-1036.
-
Hanahan D, Weinberg RA (2000) The Hallmarks of Cancer. Cell 100(1): 57-70.
-
Gatzka M, Walsh CM (2007) Apoptotic signal transduction and T cell tolerance. Autoimmunity 40(6): 442-452.
-
Kurbanov BM, Geilen CC, Fecker LF, Orfanos CE, Eberle J (2005) Efficient TRAIL-R1 / DR4-Mediated Apoptosis in Melanoma Cells by Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL). J Invest Dermatol 125(5): 1010-1019.
-
Mohana kumaran N, Hill DS, Allen JD, Haass NK (2014) Targeting the intrinsic apoptosis pathway as a strategy for melanoma therapy. Pigment Cell Melanoma Res 27(4): 525-539.
-
Lu M, Breyssens H, Salter V, Zhong S, Hu Y, et al. (2013) Restoring p53 Function in Human Melanoma Cells by Inhibiting MDM2. Cancer Cell 23(5): 618-633.
-
Krayem M, Journe F, Wiedig M, Morandini R, Najem A, et al. (2016) p53 Reactivation by PRIMA-1 Met ( APR- 246 ) sensitises BRAF melanoma to vemurafenib. Eur J Cancer 55: 98-110.
-
Panda PK, Mukhopadhyay S, Das DN, Sinha N, Naik PP, et al. (2015) Mechanism of autophagic regulation in carcinogenesis and cancer therapeutics. Semin Cell Dev Biol 39: 43-55.
-
Jones PA, Issa JJ, Baylin S (2016) Targeting the cancer epigenome for therapy. Nat Rev Genet 17(10): 630- 641.
-
Gatzka MV (2018) Targeted Tumor Therapy Remixed An Update on the Use of Small-Molecule Drugs in Combination Therapies. Cancers 10(6).
-
Derrisen EJB, Beijneh JH, Schellens JHM (2013) Concise Drug Review: Azacitidine and Decitabine. Oncologist 18(5): 619-624.
-
Suraweera A, Byrne KJO, Richard DJ (2018) Combination Therapy with Histone Deacetylase inhibitors (HDACi) for the Treatment of Cancer : Achieving the Full Therapeutic Potential of HDACi. Front Oncol 8:92.
-
Li H, Fan X, Houghton J (2007) Tumor Microenvironment : The Role of the Tumor Stroma in Cancer. J Cell Biochem 101(4): 805-815.
-
Lord CJ, Ashworth A (2012) The DNA damage response and cancer therapy. Nature 481(7381): 287-294.
-
Davar D, Beumer JH, Hameih L, Tawbi H (2012) Role of PARP Inhibitors in Cancer Biology and Therapy. Curr Med Chem 19(23): 3907-3921.
-
Mccabe N, Hanna C, Walker SM, Gonda D, Li J, et al. (2015) Mechanistic Rationale to Target PTEN- Deficient Tumor Cells with Inhibitors of the DNA Damage Response Kinase ATM. Cancer Res 75(11): 2159-2165.
-
Kumari S, Badana AK, G MM, Shailender G, Malla R (2018) Reactive Oxygen Species : A Key Constituent in Cancer Survival. Biomark Insights 13: 1177271918755391.
-
Al-jaderi Z, Maghazachi AA (2016) Utilization of Dimethyl Fumarate and Related Molecules for Treatment of Multiple Sclerosis,Cancer, and Other Diseases. Front Immunol 7: 278.
-
Nicolay JP, Karin M, Schroeder A, Brechmann M, Möbs M, et al. (2016) Dimethyl fumarate restores apoptosis sensitivity and inhibits tumor growth and metastasis in CTCL by targeting NF-kB. Blood 128(6): 805-815.
-
Guzman ML, Li X, Corbett CA, Rossi RM, Bushnell T, et al. (2007) Rapid and selective death of leukemia stem and progenitor cells induced by the, compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8). Blood 110(13): 4436-4444.
-
Mastrangelo D, Pelosi E, Castelli G, Lo coco F, Testa . (2018) Mechanisms of anti-cancer effects of ascorbate : Cytotoxic activity and epigenetic modulation. Blood Cells Mol Dis 69: 57-54.
-
Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, et al. (2014) Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508(7494): 108-112.
-
Petrachi T, Romagnani A, Albini A, Longo C, Argenziano G, et al. (2017) Therapeutic potential of the metabolic modulator phenformin in targeting the stem cell compartment in melanoma. Oncotarget 8(4): 6914-6928.
-
Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic- targeting therapy for cancer. Br J Cancer 99(7): 989- 994.
-
Rayner E, Van Gool IC, Palles C, Kearsey SE, Bosse T, et al. (2016) A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 16(2): 71-81.
-
Swanton C, Mcgranahan N, Starrett GJ, Harris RS (2015) APOBEC Enzymes : Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov 5(7): 705-712.
-
Jamal Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, et al. (2017) Tracking the Evolution of Non–Small-Cell Lung Cancer. N Engl J Med 376(22): 2109-2121.
-
Amirouchene angelozzi N, Swanton C, Bardelli A (2017) Tumor Evolution as a Therapeutic Target. Cancer Discov.
-
Skora AD, Luber BS, Azad NS, Laheru D, Diaz LA, et al. (2015) PD-1 Blockade in Tumors with Mismatch- Repair Deficiency. N Engl J Med 372(26): 2509-2520.
-
Ventura A, Kirsch DG, Mclaughlin ME, Tuveson DA, Grimm J, et al. (2007) Restoration of p53 function leads to tumour regression in vivo. Nature 445(7128): 661-665.
-
Pearson A, Smyth E, Babina LS, Herrera-abreu MT, Tarazona N, et al. (2017) High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov 6(8): 838- 851.
-
Dow LE, O Rourke KP, Simon J, Tschaharganeh DF, Van Es JH, et al. (2016) Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 161(7): 1539- 1552.
-
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, et al. (2014) Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Eng J Med 371(23): 2189-2199.
-
Dupage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T (2012) Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482 (7385): 405-409.
-
Van Den Boorn JG, Hartmann G (2013) Turning tumors into vaccines: co-opting the innate immune system. Immunity 39(1): 27-37.
-
Liu B, Song Y, Liu D (2017) Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J Hematol Oncol 10: 174.
-
Hodi FS, Day SJO, Mcdermott DF, Weber RW, Sosman JA, et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8): 711-723.
-
Bondarenko I, Rober C, Thomas L, Weber J, Garbe C, et al. (2011) Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N Eng J Med 364(26): 2517-2526.
-
Yue EW, Sparks R, Polam P, Modi D, Douty B, et al. (2017) INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med Chem Lett 8(5): 486-491.
-
Zhai L, Spranger S, Binder D, Gritsina G, Lauing KL, et al. (2016) Molecular Pathways: Targeting IDO1 and Other Tryptophan Dioxygenases for Cancer Immunotherapy. Clin Cancer Res 21(24): 5427-5433.
-
Soliman HH, Minton SE, Han HS, Ismail khan R, Neuger A, et al. (2016) A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 7(16): 22928-22938.
-
Beatty GL, Dwyer PJO, Clark J, Shi JG, Bowman KJ, et al. (2017) First-in-Human Phase 1 Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients With Advanced Solid Malignancies. Clin Cancer Res 23(13): 3269-3276.
-
Gangadhar TC, Hamid O, Smith DC, Bauer TM, Wasser JS, et al. (2015) Preliminary results from a Phase I/II study of epacadostat (incb024360) in combination with pembrolizumab in patients with selected advanced cancers. J Immunother Cancer 3(S2): O7.
-
Zhu Y, Zang Y, Zhao F, Li Z, Zhang J, et al. (2017) Inhibition of HIF-1α by PX-478 suppresses tumor growth of esophageal squamous cell cancer in vitro and in vivo. Am J Cancer Res 7(5): 1198-1212.
-
Schelman WR, Mohammed TA, Traynor AM, Kolesar JM, Marnocha RM, et al. (2014) A Phase I study of AT- 101 with Cisplatin and Etoposide in patients with advanced solid tumors with an Expanded Cohort in Extensive-Stage Small Cell Lung Cance. Invest New Drugs. 32(2): 295-302.
-
Kershaw S, Cummings J, Morris K, Tugwood J, Dive C (2015) Optimisation of immunofluorescence methods to determine MCT1 and MCT4 expression in circulating tumour cells. BMC Cancer 15:387.
-
Biology C, Tarallo V, De Falco S (2015) The vascular endothelial growth factors and receptors family : Up to now the only target for anti-angiogenesis therapy. Int J 64: 185-189.
-
Bueno MJ, Mouron S, Quintela fandino M (2017) Personalising and targeting antiangiogenic resistance : a complex and multifactorial approach. Br J Cancer (9): 1119-1125.
-
Yu Y, Cui J (2018) Present and future of cancer immunotherapy : A tumor microenvironmental perspective (Review). Oncol Lett 16(4): 4105-4113.
-
Misale S, Di Nicolantonio F, Sartore bianchi A, Siena S, Bardelli A (2014) Resistance to Anti-EGFR Therapy in Colorectal Cancer : From Heterogeneity to Convergent Evolution. Cancer Discov 4(11): 1269- 1280.
-
Place AE, Huh SJ, Polyak K (2011) The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res 13(6): 227.
-
Yu M, Bardia A, Aceto N, Bersani F, Madden MW, et al. (2014) Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345(6193): 216-220.
- Research Progress of Induced Pluripotent Stem Cells and Their Clinical Application Prospects
- Nishan Al-Kamal is the Starting Point of A Feminist Scientist
- Current Concepts and Future Perspectives of Stem Cell Therapy in Peripheral Arterial Disease
- Stem Cell and Oxidative Stress-Inflammation Cycle
- Adipose Derived Mesenchymal Stem Cells Origin, Characteristics and Promises
- Mitochondria Targeted Antioxidants can Improve In Vitro Embryo Production in Buffalo