Pregnancy Management and Outcome in ADAMTS13 Deficiency: A Case Report
<p style="text-align: justify;">Thrombotic Thrombocytopenic Purpura (TTP) is a genetic hematological disorder which includes deficiency of “Von willebrand factor cleaving protease†along with severe thrombotic microangiopathy that leads to microangipathic hemolytic anemia. Several records are taken to justify the following disease at differential age, with variegated level of severity. Case of pregnant women of 25 years is studied in this following study, her blood parameters are screened and treatments are done, on the basis of it. The proposed outcome revealed that pregnant women face much higher risk at carrying the hereditary TTP. Proper screening has to be done of all the hematological units at the time of pregnancy, psychological screening is also equally important at that time.</p>
Introduction
Thrombotic Thrombocytopenic Purpurais a very rare hereditary blood deficiency disorder of ADAMTS13 Von willebrand factor (vWf)cleaving protease and a life threatening thrombotic microangiopathy characterized by thrombocytopenia and microangipathic hemolytic anemia. The deficiency in ADAMTS 13 metalloprotease, which cleave the vWf, may be congenital or acquired. The congenital form is caused by inherited mutations in the ADAMST13 gene. The diagnosis is challenging because of nonspecific signs and symptoms, as well as the rarity of the disease. Thrombotic thrombocytopenic purpurais serious condition resulting from platelet aggregation mainly at microcirculation level, which results in occlusive ischemia [1]. Occlusion is caused by micro thrombi derived from platelet and vWf. The most common organ involved is brain. Kidneys and gastrointestinal system is less commonly affected. Deficiency causes formation of abnormally large polymer attracting platelet and formation of fibrin deposit and systemic deposition of platelet thrombi on capillary vessels and arteries. The disease usually results from a reduction in the activity of enzyme ADAMTS13, this reduction is either due to autoantibodies (Acquired TTP) or ADAMTS13 gene mutation (inherited TTP). Incidence of acquired TTP is 2.2 cases/million/year, while incidence of Congenital TTP is much lower and it represents less than 5% of all TTP cases. Incidence of inherited form of TTP is 1/1,000,000 [2, 3]. Children and neonate usually manifest familial TTP. However female may present with acute attack of the disease during pregnancy [3]. Females are more frequently affected than males, almost twice the incidence, because of the risk of acute attacks during pregnancy [4, 5]. Patient will present with sudden appearance of neurological symptoms, kidney function disturbance and fever. As inherited TTP is very rare and complicates the course of pregnancy, early diagnosis, more frequent follow up and extensive monitoring is the key to successful pregnancy outcome.
Case Presentation
Twenty five year old patient, a known case of familial TTP (ADAMTS 13 deficiency) G2P0+1 at 27+1/52 weeks of gestation presented to ER with fatigue, low appetite, vomiting and absent fetal movement. On arrival she was vitally stable except mild fever (37.8 C). A bedside scan revealed that the cardiac activity was absent which was later confirmed by an official scan. Her blood works revealed Hb 42gm/dl and platelet of 65, blood picture also revealed signs of hemolysis (increase LDH, reticulocyte count and increase indirect bilirubin). Her ADAMTS-13 activity was<0.01. ADMTS 13 antibodies were negative. She had similar episode of event during her previous pregnancy when the IUFD was diagnosed at 20 week with severe relapse of her condition, and she took one year in complete recovery thereafter. Otherwise she was medically and surgically free. She has 1 sister and 4 brothers. Sister is affected but brothers have mild hematological deficiency during early childhood which gets settled later on it’s own? Her elder sister who is 30 years old p2+1 also had congenital TTP Schulman- Upshaw Syndrome and associated severe deficiency of ADAMTS13. Her 1st pregnancy ended up IUFD at 24 weeks, baby tested positive for ADAMTS13 deficiency. Her 2nd pregnancy reached term with extensive monitoring and SSD plasma transfusion and deliver at 36 weeks. She had an early miscarriage in her last pregnancy. She had on and off admission during pregnancies for plasma and FFPs and once hospitalized for stroke during pregnancy.
Management and Outcome
G2P1 25/52 weeks, known case of Familial TTP came with c/o left upper arm weakness, numbness and chest discomfort. Patient was on regular SD plasma transfusion during pregnancy (3 units every 4 weeks) and had regular followed up with hematology in our hospital. Patient was vitally stable. Abdominal examination uterus was 25 weeks size, non-tender. On vaginal examination os was closed there was no abnormal vaginal discharge. Initial neurological assessment reveals no sensory or motor loss in the upper arm. Bilateral purpuric eruptions noted on both legs. Bedside scan revealed 25/52 weeks non-viable fetus with absent fetal heart. Initial workup done showed Hb – 5.9, platelet – 7, LDH 8, Direct Bilirubin 14.3, AST 65. Official ultrasound was requested and patient was admitted to HDU for urgent hematology and Neurology consultation. Hour after admission she started complaining of perioral numbness and mouth deviation. Neurology confirmed left sided upper arm sensory and motor involvement. There impression was presentation is likely a classic manifestation of her primary disease, however acute stroke cannot be ruled out and brain CT was requested. Hematology reviewed and started FFP transfusion 6 units bid until platelet reach 140. They advise to give 2 unit PRBcs as well. Patient labs (CBC, LDH, Reticulocyte count and RFT) repeated twice daily - Laboratory tests performed on the first day after admission and results (Tables 1-3).
| Complete Blood Count | |||
| Erythrocytes | 2.07 × 1012 cells/L | ||
| Hemoglobin | 5.9gm/dl | ||
| Hematocrit | 0.18 | ||
| White blood cells | 11.60 × 109/L | ||
| Platelets | 7 × 109/L | ||
| Reticulocyte count | 1.80% | ||
| Coagulation | |||
| PT | 10.9 sec | ||
| aPTT | 26.5 sec | ||
| INR | 1.00 g/L |
Table 1: Laboratory tests performed on the first day after admission and results.
| Other tests | Results | ||||
|---|---|---|---|---|---|
| Total bilirubin | 1.0 mg/dL | ||||
| Uric acid | 482 mg/dL | ||||
| Lactate dehydrogenase | 2482U/L | ||||
| Ferritin blood test | 148 ng/Ml | ||||
| Transferrin saturation | 11.80% | ||||
| Serum iron | 38 μg/dL |
Table 2: Blood chemistry. Patient’s clinical and laboratory parameter started improving with FFP transfusion. Brain CT done came
Table 3: Blood chemistry. Patient’s clinical and laboratory parameter started improving with FFP transfusion. Brain CT done came out normal plan from Ob Gyn team was to induce termination when platelet Count rise above 50 and hemoglobin rise above 80.Patient receive 17 units of FFP and 1 unit of PRBcs since admission. Patient started to have tachypnea and desaturation down to 82% on room air. She became oliguric as well. On examination shew had bilateral coarse crackles up to the middle zone with raised JVP. Chest x- ray done shows signs of congestion. Repeat labs Hb – 61, platelet 94 LDH – 1126, Reticulocyte count 180, Bilirubin total 28.7. ABD showed type 1 respiratory failure. Initial impression was volume overload and patient was give Furosemide 40mg IV stat. She receives another unit of PRBc with 20mg I/V Furosemide. Patient continues to DE saturate despite oxygen therapy and shifted to ICU for respiratory support. Patient condition deteriorated clinically and she was intubated (NIV). Impression from ICU team: Anaphylactic reactions to blood products with volume overloadvs Septic abortion Termination of pregnancy was started with tab Misoprostol and she aborted completely after single dose Official scan confirmed empty uterine cavity. Histopathology reveals placenta with intervillous fibrin infarcts and calcification. Her clinical and laboratory parameter improved remarkably after termination, she was extubated the same day. Her labs CBC 78, PLT 188, LDH 614, ALT 45, AST 17 Shifted to the ward, seen by hematology and plan was to transfuse SD plasma 4 units every 3-4 Weeks and OPD follow up after 1 week.
Discharge in good asymptomatic condition while her Hemoglobin was 10.6 and PLT were 388. UN remarkable OPD follow up done with OB/GY after 2 week.
Discussion
We are reporting from king Abdul-Aziz Medical city Riyadh KSA. A 25 years old Patient who has acute exacerbation of her clinical condition and IUFD twice in the context of Familial Thrombotic Thrombocytopenic purpura, who despite previous diagnosis and regular monitoring by Hematology has undesirable outcome of pregnancy. Non immune mediated hereditary form of TTP or Upshaw-Schulman syndromeis rare with only 100 cases described worldwide. It is caused by mutation in the ADAMST13 gene (9q34), encoding ADAMTS13, a metalloprotease involved in the cleavage of ultra-large vWfmultimers with a penetrance of over 90%. Early TTP in childhood represents less <10% of TTP cases. The initial episode of inherited TTP occurs during early neonatal period or childhood representing physiological stress of the disease at that time. Disease severity may vary from isolated thrombocytopenia and isolated billirubinemia. Disease severity is variable depending upon specific mutation of ADAMST13 [1]. ADAMTS 13 deficiencies alone are not sufficient to cause TTP. Environmental factors may contribute to induce full blown manifestation of the disease. Potential triggering factors include pregnancy and infection. When associated with pregnancy most cases occur during second half of the pregnancy like in this patient and are associated with USS (Upshaw–Schulman syndrome) along with hypercoagulable state with ADAMST 13 deficiency [3]. Differential diagnosis include 1-HUS (hemolytic uremic syndrome), 2-ITP (Immune thrombocytopenic purpura), 3-HELLP syndrome 4-Acquired TTP. Hemolytic Uremic syndrome Hemolytic uremic syndrome (HUS) is a condition characterized by destruction of red blood cells, low platelet count, and kidney failure. Adults with atypical HUS tend to become more ill and need more aggressive therapy than children with the condition. In addition to the supportive care discussed above, plasmapheresis or plasma exchange may be required (Table 4).
| Clinical and Laboratory Features | TTP | HUS | HELLP | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Neurologic s/s | +++ | +/- | +/- | ||||||||
| Fever | ++ | +/- | - | ||||||||
| Hypertension | +/- | +/- | +/- | ||||||||
| Renal dysfunction | +/- | +++ | +/- | ||||||||
| Skin lesions-purpura | + | - | - | ||||||||
| Platelets | ↓ ↓ ↓ | ↓ ↓ | ↓ | ||||||||
| Prothrombin time (PT)/activated partial thromboplastin time (aPTT) | ⇔ | ⇔ | ↑ or ⇔ | ||||||||
| Fibrinogen | ⇔ | ⇔ | ↑ or ⇔ | ||||||||
| BUN/creatinine | ↑ | ↑ ↑ ↑ | ↑ or ⇔ | ||||||||
| Aspartate aminotransferase (AST)/alanine aminotransferase (ALT) | ⇔ | ⇔ | ↑ | ||||||||
| Lactate dehydrogenase (LDH) | ↑ | ↑ ↑ ↑ | ↑ |
Table 3: Differential diagnosis. ITP is not associated with microangiopathic changes on the peripheral blood smear, such as absen
Table 4: Differential diagnosis. ITP is not associated with microangiopathic changes on the peripheral blood smear, such as absence of schistocytes and will not cause renal or neurological abnormalities. ADAMTS13 level in the blood will be normal as well [6]. HELLP syndrome (hemolyticanemia, elevated liver enzyme, and low platelet count) occurs in association with Hypertension associated with Preeclampsia and leads to multiorgan failure [7]. Both of the diseases share similar clinical and laboratory parameter and it is difficult to differentiate. Both associated with thrombocytopenia and microangiopathic changes on peripheral blood smear [8]. Differentiating HELLP syndrome with TTP is occasionally possible when abnormalities persist after delivery [9]. HELLP syndrome did not associated with ADAMST13 deficiency and requires treatment of underlying syndrome such as delivery rather than plasma transfusion. Acquired TTP is more common than inherited TTP. Both are characterized by thrombocytopenia and microangiopathic hemolytic anemia and may represent with renal and neurological abnormalities. They are caused by severely deficient activity of vWf protease ADAMST13 (<10%). However acquired TTP is caused by autoantibodies which can be detected in the blood. Familial TTP occur early and in first pregnancy. Fetal loss rate is around 40% [10]. Plasma infusion normally corrects ADAMST13 level and improves symptoms. Immunosuppressive therapy is unnecessary. The possibility of hereditary TTP should be considered in patient who present with microangiopathic hemolytic anemia and thrombocytopenia , peripheral smear shows schistocytes and low platelet count and low level of ADAMST13 deficient (<10%). Acute exacerbation must be treated by plasma infusion (10-15ml/kg/day) [11]. In resistant cases plasma exchange may be required. Patient with chronic relapsing disease should be considering for regular plasma infusion to maintain ADAMST13 activity above 15% [12]. Mortality rate without treatment is about 90% [13]. With plasma exchange and plasma infusion a decrease in mortality rate to around 15% [14, 15, 16, 17] (Figure 1).
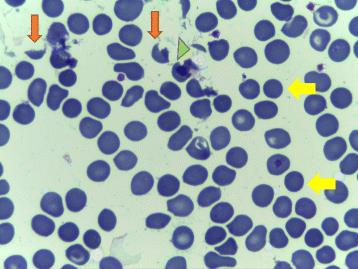
Conclusion
Patient with hereditary TTP are high risk pregnant population. They should have appropriate pre pregnancy counselling regarding management of TTP before and during pregnancy. There should be regular follow up of plasma infusion and laboratory checks. During pregnancy it should be multidisciplinary approach with involvement of neurology, nephrology and OBGYN.
Refferences
1. Joly BS, Coppo P, Veyradier A (2017) Thrombotic
thrombocytopenic purpura. Blood 129: 2836-2846.
2. Reese JA, Muthurajah DS, Kremer Hovinga JA, Vesely
SK, Terrell DR, et al. (2013) Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: comparison of incidence, demographic and clinical features. Pediatr Blood Cancer 60(10): 1676-1682.
3. Moatti-Cohen M, Garrec C, Wolf M, Boisseau P,
Galicier L, et al. (2012) Unexpected frequency of Upshaw-Schulman syndrome in pregnancy-onset thrombotic thrombocytopenic purpura. Blood 119(24): 5888-5897.
4. Török TJ, Holman RC, Chorba TL (1995) Increasing
mortality from thrombotic thrombocytopenic purpura in the United States—analysis of national mortality data, 1968–1991. Am J Hematol 50(2): 84- 90.
5. Fujimura Y, Matsumoto M, Isonishi A, Yagi H, Kokame
K, et al. (2011) Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost 9(1): 283-301.
6. Quintero V, Garcia-Pose A, Barrios-Tascon A, Pacheco-Cumani M (2014) Congenital ADAMTS13 deficiency: a rare mimicker of immune thrombocytopenic purpura. J Pediatr Hematol Oncol 36(8): 653-655.
7. Martin JN, Rose CH, Briery CM (2006) Understanding
and managing HELLP syndrome: The integral role of aggressive glucocorticoids for mother and child. Am J Obstet Gynecol 195(4): 914-934.
8. Matsumoto M, Fujimura Y, Wada H, Kokame K,
Miyakawa Y, et al. (2017) Diagnostic and treatment guidelines for thrombotic thrombocytopenic purpura (TTP) 2017 in Japan. Int J Hematol 106(1): 3-15.
9. Townsley DM (2013) Hematologic complications of pregnancy. Semin Hematol 50(3): 222-231.
10. Knöbl P (2014) Inherited and acquired thrombotic thrombocytopenic purpura (TTP) in adults. SeminThromb Hemost 40(4): 493-502.
11. Lotta LA, Garagiola I, Palla R, Cairo A, Peyvandi F
(2010) ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum Mutat 31(1): 11-19.
12. Barbot J, Costa E, Guerra M, Barreirinho MS, Isvarlal
P, et al. (2001) Ten years of prophylactic treatment with fresh-frozen plasma in a child with chronic relapsing thrombotic thrombocytopenic purpura as a result of a congenital deficiency of von Willebrand factor-cleaving protease. Br J Haematol 113(3): 649- 651.
13. Scully M, Hunt BJ, Benjamin S, Liesner R, Rose P, et al.
(2012) Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 158(3): 323-335.
14. Tărniceriu CC, Mircea-Vicol R, Anton E, Ancuţa C,
Bădulescu OV, et al. (2014) Thrombotic thrombocytopenic purpura: A hematological emergency. Rom J Morphol Embryol 55(3): 1259- 1262.
15. Chauhan AK, Motto DG, Lamb CB, Bergmeier W,
Dockal M, et al. (2006) Systemic antithrombotic effects of ADAMTS13. J Exp Med 203(3): 767-776.
16. Lúcio D de S, Pignatari JF, Cliquet MG, Korkes HA
(2017) Relapse of congenital thrombotic thrombocytopenic purpura, after spontaneous remission, in a second-trimester primigravida: case report and review of the literature. Sao Paulo Med J 135(5): 491-496.
17. Semple JW (2002) Immune pathophysiology of autoimmune thrombocytopenic purpura. Blood Rev 16(1): 9-12.
- Psychogenic Erectile Dysfunction in Late Adulthood: A Case Report on Clinical Intervention and Intimacy Restoration
- Clinical Trials on COVID-19 in 2025: A New Chapter in Global Health Research
- Innovations and Challenges in Contemporary Medical Clinical Trials: An Editorial Perspective
- Innovations and Challenges in Contemporary Medical Clinical Trials: A Critical Perspective
- Reimagining Clinical Trials: The Power of Continuous Feedback from Medical Reports
- Factors Influencing Brain Drain: Perspectives from a Medical School in Turkey