Unusual Association of Two Germline Rearrangements in CDC73/HRPT2 Gene: Complexity of Clinical Counseling in the Age of Genomic Medicine
Background: Primary hyperparathyroidism is a phenotype shared by several hereditary endocrine and non-endocrine cancer predisposition syndromes caused by heterozygous germline mutations deactivating tumor suppressor genes including CDC73 (=HRPT2). Objectives and Results: We describe the genetic investigation of a family in which the proband was a 16- year-old male patient suffering from primary hyperparathyroidism related to a unique parathyroid adenoma. His DNA analyses revealed a CDC73 germline heterozygous deletion of exons 7 to 13 in the 1q31.2 locus of the chromosome 1. Genetic investigations in his parents who were both asymptomatic showed that the father was not carrying the deletion. Two chromosomal rearrangements were carried by the mother, both affecting CDC73: the deletion previously identified in her son and a 626 kb duplication encompassing 6 contiguous genes: UCHL5, TROVE2 GLRX2, CDC73, MIR1278 and B3GALT2. This duplicated segment was inserted into chromosome 20. Lack of symptoms in the probands mother suggests that the CDC73 extra copy is able to balance the loss of function caused by the deleted allele by allowing a normal level of parafibromin expression. Conclusion: For the first time, we describe the association of two large and complex genetic events in CDC73 gene complicating genetic counseling. Literature doesnt provide much information about potential consequences of these genes deletion or duplication, thus highlighting the importance of multidisciplinarity in the practice of precision and genomic medicine. Moreover, this study demonstrates that copy number variations should be carefully investigated in routine diagnosis in order to deliver an accurate genetic counseling to those families.
Introduction
Hyperparathyroidism-jaw tumor syndrome (HPT-JT), familial isolated hyperparathyroidism (FIHP) and parathyroid carcinoma are three phenotypes belonging to the spectrum of CDC73-related disorders. They all arise from structural variations that deactivate the tumor suppressor gene CDC73 (also known as HRPT2, NM_024529.4). Diagnosis is based on the biochemical findings of primary hyperparathyroidism (PHPT), imaging studies, family history and detection of a heterozygous germline CDC73 pathogenic variant. CDC73- related disorders display autosomal dominant inheritance with variable penetrance, the proportion of cases caused by de novo pathogenic variant being still unknown [1, 2]. The CDC73 gene, located to the 1q31.2 locus, comprises 17 exons, and encodes parafibromin, a component of the RNA polymerase II and PAF1 complex involved in transcriptional regulation and histone modifications [3]. CDC73 pathogenic variations are mostly frame-shift or nonsense mutations leading to the occurrence of a truncated parafibromin [4]. In the absence of point mutations, CDC73 intragenic deletions have to be searched in cases of high risk patients [5]. Here, we describe in a family two chromosomal rearrangements both affecting CDC73: a 7 exons deletion and a duplication of 6 genes including CDC73.
Case Report and Results
A 16 years old boy was initially referred for kyphosis investigations. Radiological screening incidentally detected osteolytic lesions of 35 mm in left femur and of 20 mm in left greater trochanter. Biological tests (Figure 1A) revealed primary hyperparathyroidism. Echography detected a hypervascular left parathyroid nodule (15x15x20 mm) and technetium bone scan an osteolytic area in the left trochanter corresponding to a brown tumor (Figure 1B). Anatomopathological analysis after parathyroid surgery (February 2010) confirmed the presence of a unique parathyroid adenoma with oncocytic cells. There was no familial history of endocrinopathy and his parents and sister were normocalcemic. In front of the early-onset of this PHPT, molecular genetic studies were performed after collection of informed consent. CDC73 exonic regions and their flanking junctions were fully sequenced by Sanger sequencing [5] but no mutation was detected. Further analyses by qPCR (Taqman) and MLPA (P466-A1 kit, MRC Holland) (Figure 2A) left revealed a heterozygous deletion of exons 7 to 13. Moreover, according to the Knudson model, a second allele inactivation in the tumor was characterized (exon 2: c.142G>T, p.(Glu48*)). Thus, complete loss of function of CDC73 was ascertained. Genetic investigations were performed in the kindred to discriminate de novo from familial mutation and to provide genetic counseling. DNAs from proband’s father, mother and sister were analyzed by MLPA and qPCR. No deletion was found in father and sister. In proband’s mother, 3 copies for exons 1 to 6 and 14 to 17 of CDC73 gene were detected (Figure 2A) right suggesting an additional event. Comparative Genomic Hybridation (array-CGH) analysis (60K oligo probes (Agilent)) was performed in the mother and found a CDC73 heterozygous deletion of exons 7 to 13 (80 kb;
chr1:193,110,996-193,190,959; GRCh37/hg19) associated with a heterozygous duplication encompassing CDC73 and 5 contiguous genes (UCHL5, TROVE2, GLRX2, MIR1278, B3GALT2 ; 626 kb: chr1:192,805,274- 193,431,181; GRCh37/hg19) (Figure 2B). FISH analysis located this extra-copy inserted into chromosome 20.
| 250H Vitamin D | Phosphatemia | Calciuria mmol/ | |||||||||||||||
| Date | Calcemia mmol/L | PTH pg/ml | |||||||||||||||
| ng/ml | mmol/L | 24h | |||||||||||||||
| Normal ranges | 2,18-2,58 | 11-54 | 26-65 | 0,8-1,5 | 2, 5-6,2 | ||||||||||||
| December-09 | 4,2 | 870 | 0,58 | 12 | |||||||||||||
| February 2010 | 2,12 | 8 | |||||||||||||||
| July 10 | 2,37 | 22 | 17 | ||||||||||||||
| September 10 | 2,48 | 44 | |||||||||||||||
| January 12 | 2,41 | 19,5 | 22,4 |
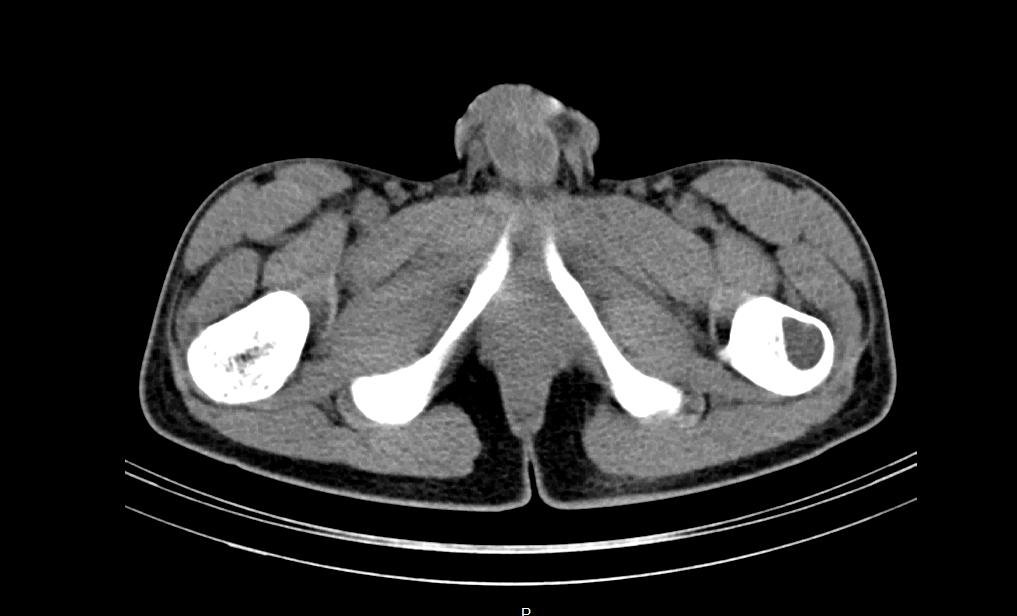
1B: Technetium bone scan (January 2010) detected the presence of an osteolytic area in the left femur (white arrow).
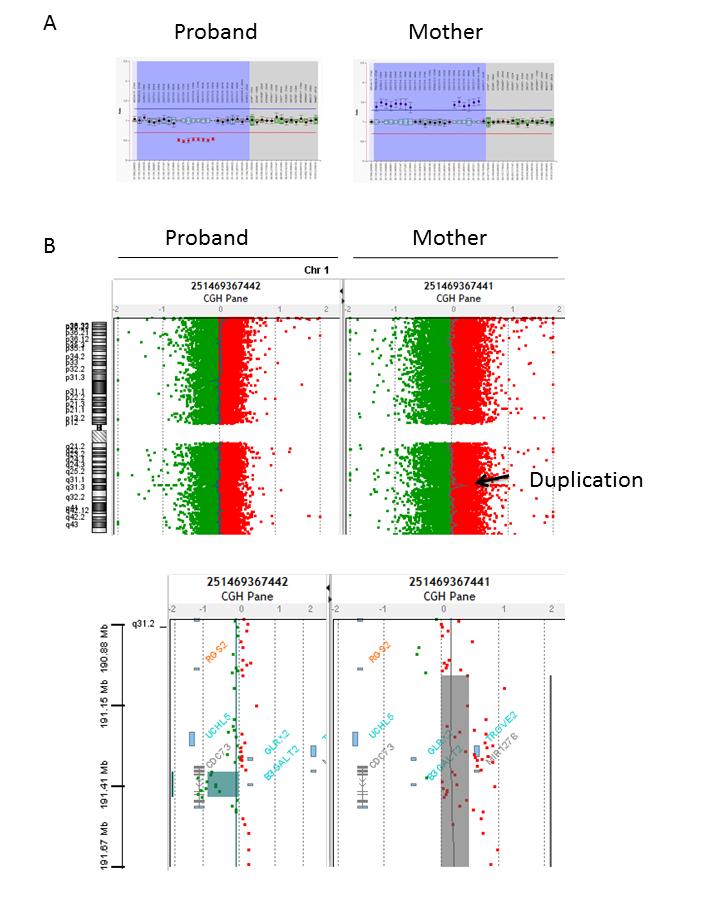
2A: “Salsa MLPA probemix P466-A1 CDC73” kit (MRC Holland). Each box plot corresponds to a probe hydrization on each CDC73 exon (blue area) in addition to control probes on other chromosomes on the right (grey area). Normal hybridization ratio is comprised between the two horizontal bars (mean ± 2sd, blue and red bars). A signal with a ratio around 0.5 suggests a heterozygous deletion and a ratio around 1.5 suggests a heterozygous duplication.
2B: Array CGH analysis of the proband and his mother’s DNA (Agilent oligonucleotide array 60K; up: complete view of the chromosome, Bottom: zoom on the affected region). The right panel highlights the duplicated region in the mother DNA whereas the left zoom shows the deletion of exons 7 to 13 in the proband’s DNA.
Discussion
Four other cases of partial or whole CDC73 deletion have also been described in literature [5, 6, 7, 8, 9, 10]. So CDC73 gross deletions are not that uncommon, with an estimated frequency around 7% [5]. Interestingly, one study [10] described in 1q32 locus a large germline deletion affecting the 6 contiguous genes we found duplicated in the proband’s mother DNA. Indeed, literature highlights the inherent potential of this region, corresponding to a conserved haplotype block structure across mammals [11], to be involved in breakage and recombination events. Large duplications mostly result in direct tandem or interstitial insertion mechanism [12]. In our study, insertion into chromosome 20 was quite unexpected but explains the familial segregation of the disease, especially the absence of CDC73 mutated allele in sister’s proband for chromosome 1 and 20. These results raised the issue of the consequence of the extra CDC73 gene copy, the proband’s mother being still asymptomatic at the age of 56. Haploinsufficiency resulting from the partially deleted allele might be properly balanced by the CDC73 extra- copy. However, annual phosphocalcic metabolism monitoring and cervical echography every 2 to 3 years were recommended, the long term consequences of the association of such unusual events remaining unknown. Here, four modes of chromosomal segregation could be expected and have to be explained during genetic counseling, as well as their potential functional consequences: a. Apart from genotypes identified in the proband and his mother, we could observe a complete normal profile for the offspring or duplication of the 626 kb locus without the 80 kb segmental deletion. b. The pathogenicity of most duplications is not only explained by an extra-copy of one or several genes leading to triplosensitivity but by disruption or misregulation of genes that span duplication breakpoints. c. In addition to CDC73, 5 other genes are involved in the proband’s mother rearrangement: UCHL5, GLRX2, TROVE2, MIR1278 (3 copies) and B3GALT2, an overlapping CDC73 gene in intron 10 (2 copies). Large CNV, larger than 400 or 500 kb, have been described in control populations [13, 14], but both gene density and size are strongly anticorrelated with allele frequency. Thus, size alone can’t be used as a predictor of pathogenicity [13]. The 626 kb CNV identified in our study wasn’t reported in literature thus leading us to search for diseases individually associated with these genes. UCHL5 encodes a deubiquitinating enzyme, GLRX2 a glutathione-dependant oxydoreductase involved in oxydative stress response, TROVE2 a RNA-chaperone involved in congenital heart block and Sjogren’s syndrome, MIR1278 encodes a microRNA expected to be implicated in expression regulation of various genes and B3GALT2 a beta-1,3-galactosyltransferase involved in glycoprotein and glycolipid biosynthesis. No particular diseases are reported in databases as typically associated with these genes. Interestingly, B3GALT2 overlaps CDC73 gene in intron 10 and is included in the proband’s deleted region. Cascon, et al. [10], who found all these genes deleted in one patient without troubles out of HPT, suggested that they are haplo sufficient. This hypothesis is in agreement with the absence of additional symptom for the proband and his mother. CDC73 is considered as a medically actionable gene [15], involving that incidental events found in this gene during exome analyses have to be reported. Our work highlights the difficulties of precision medicine which will be amplified in the coming years by growing number of whole exome and genome sequencing studies. It thus raises the issue of genetic counseling complexity for these incidental events not yet reported in publications and emphasizes the importance of multidisciplinary support. Literature should collect such data about these complex genetic events to create a medical bank [16, 17], as described by Aronson, et al. [18], to which refer to regarding the patient support, genetic counseling and medical monitoring of associated pathologies. In conclusion, our work describes for the first time the association of two genetic rearrangements affecting CDC73 gene. This case report highlights the importance of cooperation between the patient, his family and the medical multidisciplinary team in the age of precision medicine.
Acknowledgments
We are grateful to the patient, his sister and his parents for their cooperation and participation in this study. We thank Pascal Plouvier, Marthe Cochard, Isabelle Szuster, Wassila Igoudjil, Evelyne Destailleur, Evelyne Crème and Mélanie Schneiter for excellent technical support. We thank INCa for financial support and TENgen network for helpful discussions.
References
-
Duan K, Gomez Hernandez K, Mete O (2015) Clinicopathological correlates of hyperparathyroidism. J Clin Pathol 68(10): 771-787.
-
Thakker RV (2016) Genetics of parathyroid tumours. J Intern Med 280(6): 574-583.
-
Newey PJ, Bowl MR, Thakker RV (2009) Parafibromin--functional insights. J Intern Med 266(1): 84-98.
-
Newey PJ, Bowl MR, Cranston T, Thakker RV (2010) Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat 31(3): 295-307.
-
Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, et al. (2013) Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab 98(2): E403-408.
-
Kong J, Wang O, Nie M, Shi J, Hu Y, et al. (2014) Familial isolated primary hyperparathyroidism/hyperparathyroidism-jaw tumour syndrome caused by germline gross deletion or point mutations of CDC73 gene in Chinese. Clin Endocrinol 81(2): 222-230.
-
Korpi-Hyövälti E, Cranston T, Ryhänen E, Arola J, Aittomäki K, et al. (2014) CDC73 intragenic deletion in familial primary hyperparathyroidism associated with parathyroid carcinoma. J Clin Endocrinol Metab 99(9): 3044-3048.
-
Domingues R, Tomaz RA, Martins C, Nunes C, Bugalho MJ, et al. (2012) Identification of the first germline HRPT2 whole-gene deletion in a patient with primary hyperparathyroidism. Clin Endocrinol 76(1): 33-38.
-
Mehta A, Patel D, Rosenberg A, Myriem B, Ryan JE, et al. (2014) Hyperparathyroidism-jaw tumor syndrome: Results of operative management. Surgery 156(6): 1315-1324.
-
Cascón A, Huarte-Mendicoa CV, Javier Leandro-García L, Letón R, Suela J, et al. (2011) Detection of the first gross CDC73 germline deletion in an HPT-JT syndrome family. Genes Chromosomes Cancer 50(11): 922-929.
-
Guryev V, Smits BM, Van de Belt J, Verheul M, Hubner N, et al. (2006) Haplotype block structure is conserved across mammals. PLoS Genet 2(7): e121.
-
Newman S, Hermetz KE, Weckselblatt B, Rudd MK (2015) Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet 96(2): 208-220.
-
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, et al. (2009) Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet 84(2): 148-161.
-
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, et al. (2011) A copy number variation morbidity map of developmental delay. Nat Genet 43(9): 838-846.
-
Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, et al. (2013) Actionable, pathogenic incidental findings in 1,000 participants' exomes. Am J Hum Genet 93(4): 631-640.
-
Cook MC (2015) Medical case reports in the age of genomic medicine Clin Transl Immunology 4(10): e45.
-
Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, et al. (2015) Clin Gen--the Clinical Genome Resource. N Engl J Med 372(23): 2235-2242.
-
Aronson SJ, Rehm HL (2015) Building the foundation for genomics in precision medicine. Nature 526(7573): 336-342.
- Shaping Healthy Futures: Pediatric Endocrine Breakthroughs of 2025
- Precision Medicine in Obesity: Customizing Treatment for 2025
- The Thyroid Revolution: How 2025 is Redefining Hormone Health
- Editorial- Targeting Immunometabolism for Generating Innovative Therapies for Cancer
- Current Knowledge of Chickenpox
- Correlation of Preinjection Values of Gonadotropins and Estradiol Level with Clinical and Radiologic Evidence of Sufficient Pubertal Suppression in Girls with Central Precocious Puberty