Congenital Hyperinsulinism: Report of a New Mutation
Congenital hyperinsulinism is a rare condition characterized by inappropriate secretion of insulin leading to recurrent and persistent hypoglycaemia, which may subsequently lead to permanent brain damage. Genetic mutations have been identified in approximately 50% of cases attributable to defects in at least 11 genes. The clinical presentation is non-specific. Seizures during infancy or pallor, diaphoresis and tachycardia, in childhood, can be the first presentation. Medical treatment includes diazoxide, chlorthiazide, nifedipine and octreotide. In diazoxide unresponsive patients, further investigations including genetic studies and 18F-DOP PET scan may be warranted to identify the two distinct types of congenital hyperinsulinism described in the literature: focal and diffuse forms. Focal hyperinsulinism may be cured by excision of the focal lesion; whereas, diffuse hyperinsulinism may require near-total pancreatectomy.</p> <p>We report a female newborn with focal hyperinsulinism but with multifocal lesions. This is rarely described in the current literature. The genetic mutation identified in our case was ABCC8c.1792C>Tp.Arg598* of paternal origin, a new mutation, not previously described in the literature. Due to the multifocal nature of this form of hyperinsulinism, the patient developed recurrent episodes of hypoglycaemia despite excision of the focal lesion. Further genetic studies may be necessary in the identification of possible genetic mutations associated with multifocal hyperinsulinism, which may aid the management and predict the prognosis
Introduction
Congenital hyperinsulinism (CH) is a condition characterized by severe and persistent hypoglycaemia due to abnormal secretion of insulin. Glucose is the main energy source of the brain. In the newborn developing brain, recurrent and prolonged episodes of hypoglycaemia may lead to seizures, coma and permanent neurological sequelae; therefore, early recognition and prompt management is crucial in avoiding long term consequences. CH affects 1 in 40,000 to 50,000 live births in the general population and 1 in 2500 in consanguineous parents [1].
Pathophysiology
Hyperinsulinism hypoglycaemia may be transient, persistent or syndrome related. Transient hyperinsulinism is often secondary to perinatal causes (e.g. infant of a diabetic mother, intrauterine growth restriction) and usually settles within days after delivery and would rarely go beyond months. Syndrome related hyperinsulinemic hypoglycaemia is associated with a number of overgrowth syndromes (e.g. Beckwith- Wiedemann, Perlman, Sotos). Persistent hyperinsulinism is seen commonly in congenital hyperinsulinism. Congenital hyperinsulinism is the commonest cause of persistent hypoglycaemia in infants. Many cases of CI arise in patients without a family history of the disease. Genetic mutations have been identified in approximately 50% of cases attributable to defects in at least 11 genes [2] (Table 1). These genetic mutations may lead to channelopathies, enzyme anomalies and transcription factor defects (Table 2). Channelopathies refer to pancreatic B-cell ATP-sensitive potassium channels (KATP channels) defects. The KATP channel consists of two subunits: the sulfonylurea receptor (SUR1) which is encoded by the ABCC8 gene (responsible for 50-60% of CH cases) and the inwardly rectifying potassium channel (KIR6.2) unit which is encoded by the KCNJ11 gene (responsible for 10-15% of CH cases). It is noted that paternally inherited mutations of the ABCC8 or KCNJ11 gene and a concomitant loss of maternal allele results in focal pancreatic lesions with one or multiple focuses. When mutations of the ABCC8 or KCNJ11 gene are germ- line mutations it results in diffuse pancreatic lesions. These mutations lead to abnormal gene expression and cell growth. Enzyme anomalies and transcription factor defects may also lead to congenital hyperinsulinism but less frequently. These mutations may lead to intracellular accumulation of intermediary metabolites or altered concentration of signaling molecules subsequently causing functional abnormalities of B-cells and inappropriately high insulin secretion during hypoglycaemic episodes [3, 4, 5].
| Genes | Inheritance | Diazoxide Response | ||||||
| ABCC8 alone | dominant | - | ||||||
| ABCC8/ KCNJ11 | recessive | - | ||||||
| ABCC8/ KCNJ11 | dominant | + | ||||||
| ABCC8/ KCNJ11 | sporadic | - | ||||||
| GLUD1 | dominant | + | ||||||
| GCK | dominant | - | ||||||
| HK1 | dominant | + | ||||||
| HADH1 | recessive | + | ||||||
| HNF1A | dominant | + | ||||||
| HNF4A | dominant | + | ||||||
| SLC16A1 | dominant | - | ||||||
| UCP2 | dominant | + | ||||||
| PGM1 | recessive | - |
Table 1: Genes inheritance and diazoxide response in congenital hyperinsulinism.
| Genes | Encoding Protein | ||||
|---|---|---|---|---|---|
| Channelopathies | |||||
| ABCC8 | SUR1 subunit of pancreatic B-cell ATP-sensitive potassium channels (KATP channels) | ||||
| KCNJ11 | KIR6.2 subunit of KATP channels | ||||
| Enzyme anomalies | |||||
| GLUD1 | Glutamate dehydrogenase | ||||
| GCK | Glucokinase | ||||
| HADHSC | Short-chain L-3-hydroxyacyl-CoA dehydrogenase (SCHAD) | ||||
| Transcription factor defect | |||||
| HNF4A | Hepatocyte nuclear factor 4a | ||||
| UCP2 | Mitochondrial uncoupling protein 2 | ||||
| Insulin receptor |
Table 2: Genes involved in congenital hyperinsulinism.
hypoglycaemic episodes are often severe during both fasting and post-prandial phase requiring high concentrations of glucose infusion (>8mg/kg/h). Presenting symptoms vary and depends on the patient’s age and severity, ranging from asymptomatic to life threatening hypoglycaemic coma or status epilepticus [5, 6].
Clinical Features
Hypoglycaemia is the main presentation of congenital hyperinsulinism and is associated with high risks of seizures and brain damage. The clinical presentation of CH is most severe in newborns and may be quite subtle during infancy and childhood (Table 3). The
| Neonatal Period | Infancy and Childhood | Syndrome related CH | |||||||
| Clinical presentation | Seizures (50%) | Before 1y: | Syndrome related dysmorphic features (e.g. developmental delay, heart defects or malformations) | ||||||
| Macrosomia | Seizures | ||||||||
| Lethargy | Drowsiness | ||||||||
| Poor feeding | Excitability | ||||||||
| Apnea | |||||||||
| Coma | After1y: | ||||||||
| Mild hepatomegaly | Pallor | ||||||||
| Faint | |||||||||
| Tachycardia | |||||||||
| Sweating | |||||||||
| Seizures | |||||||||
| Response to diazoxide | Non-responsive | Responsive |
Table 3: Clinical features of hyperinsulinism
Diagnosis
Early diagnosis is crucial in preventing hypoglycaemia related permanent brain injury; therefore a high index of suspicion is necessary. Any patient with recurrent or persistent hypoglycaemia requires further investigation. (Table 4) High intravenous glucose requirement to maintain normoglycaemia is also an important telltale sign. Biochemically, hyperinsulinimic hypoglycaemia can be diagnosed by the presence of inappropriate insulin and low ketones in a hypoglycaemic state, responsiveness to glucagon and high concentration of dextrose infusion to maintain normoglycaemia (Table 5).
- Management
- Hormone
- Expected findings in hyperinsulinism
- Glucose
- Low
- Insulin
- Detectable/ raised
- C-peptide
- Detectable/ raised
- Betahydroxybutyrate
- Suppressed/ low
- Free fatty acids
- Suppressed/ low
- IGFBP-1
- Suppressed/ low
Table 4: Expected hormonal findings in hyperinsulinism
Diagnostic features of hyperinsulinism
Blood glucose <2.5mmol/L with: Inappropriate serum insulin (>1uU/mL)
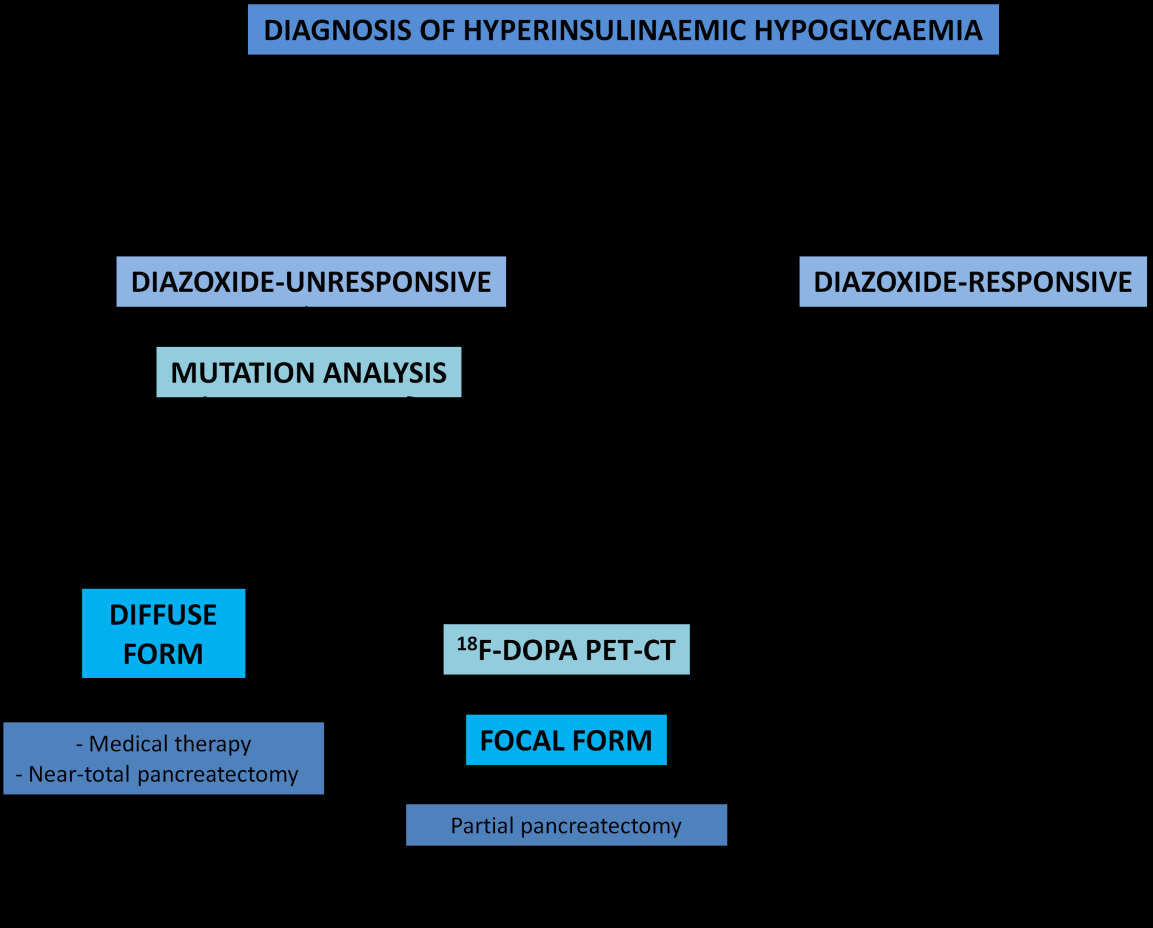
Suppressed/ low ketone bodies Suppressed/ low serum fatty acids Suppressed branch chain amino acids Positive glycaemic (>1.5mmol/L) response to im/ iv glucagon Dextrose infusion rate >8mg/kg/min to maintain normoglycaemia Table 5: Diagnostic biochemical features of hyperinsulinism Early diagnosis and prompt management to main normoglycaemia (3.5- 6mmol/L) is crucial in preventing brain injury in patients with congenital hyperinsulinism [7]. The main drugs used in CHI include diazoxide, somatostatin analogues (octreotide) (Table 6). After the initial treatment, the assessment of the response to treatment is crucial in guiding our next step in management. The first and mainstay of treatment for hyperinsulinism is diazoxide. Diazoxide is effective in all forms of hyperinsulinism except in focal congenital hyperinsulinism and diffuse congenital hyperinsulinism caused by mutations in ABCC8 and KCNJ11 that causes inactivation. The management of diazoxide unresponsive hyperinsulinism has improved in the last decade due to recent advances in molecular genetics, radiology imaging techniques and laparoscopic surgery [8]. Therefore, hyperinsulinism that is diazoxide unresponsive, further investigations should be performed looking for focal or diffuse hyperinsulinism as their surgical management differ immensely (Figure1). Focal hyperinsulinism may lead to total ‘cure’ by respecting the lesion; whereas diffuse hyperinsulinism may require near-total pancreatectomy by removing 95-98% of the pancreas for clinical improvement. Genetic studies found that patients with ABCC8 and KCNJ11 homozygous or compound heterozygous mutations are associated with diffuse disease; whereas paternal mutation in ABCC8 and KCNJ11 are associated with focal disease and therefore subsequent imaging with 18F-DOPA-PET scan is warranted to aid preoperative localization of the focal lesion. Studies showed that the sensitivity of this scan is between 88-94% and the specificity is 99-100% [3, 4, 5]. Once identification of the focal form of the disease is done, a lesionectomy or partial pancreatectomy of the affected area may lead to cure of the disease. This may be performed laparoscopically (particularly lesions in the tail of the pancreas) with less postoperative complications and faster recovery time. Lesions that are present in the head of the pancreas may require a more traditional approach of open surgery. Diffuse form of the disease requires a near-total pancreatectomy which is associated with higher risk of developing diabetes later on in life; therefore, should be considered only in severe cases that have been unresponsive to all medical therapy.
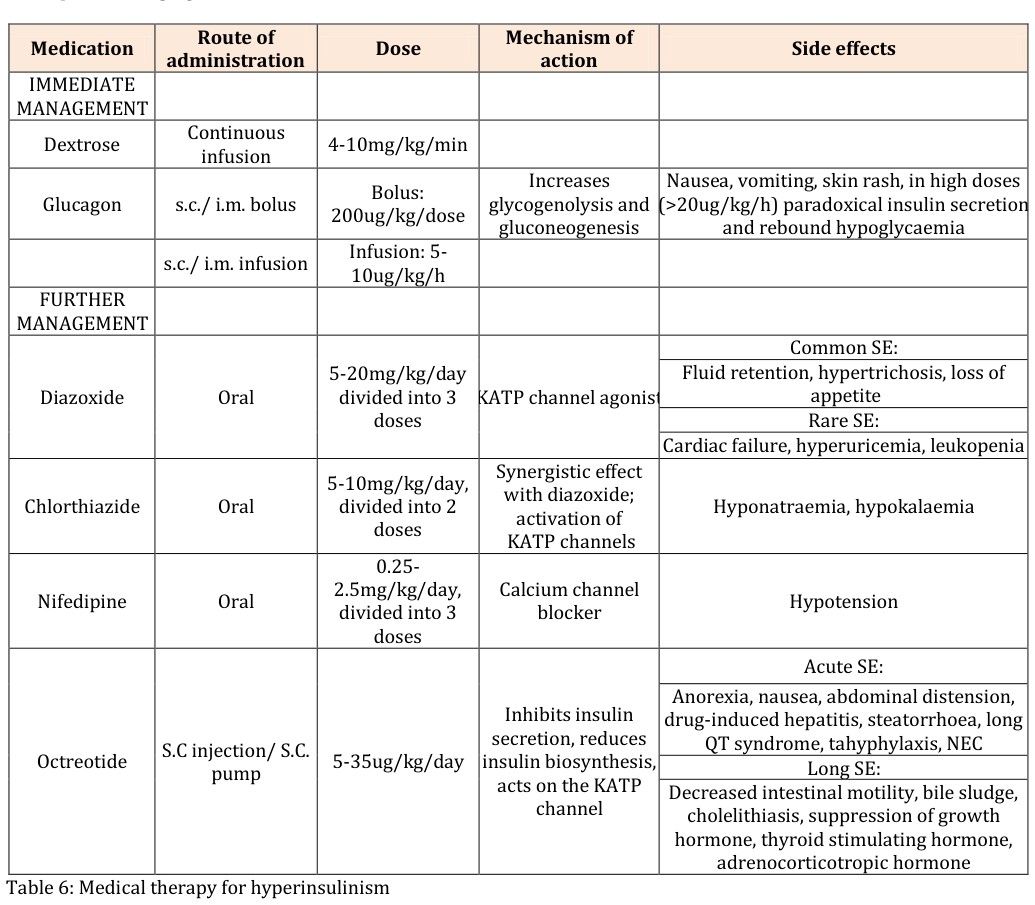
| hormone, thyroid stimulating hormone, adrenocorticotropic hormone |
Table 5: Medical therapy for hyperinsulinism
Case Report
We describe a case of a female newborn with a normal perinatal history (delivery via caesarean section at 37 weeks and 4 days of gestation due to large for gestational age and cephalopelvic disproportion. Birth weight was 4.3kg (>95th centile), height of 52cm (75-90th centile) and head circumference of 35cm (50-75th centile). Capillary blood glucose performed prior to first feed at 2 hours of age showed hypoglycaemia (1.0mmol/L). Urine ketone and reducing substances were negative. Lactate, arterial blood gas, metabolic and hormonal screen were unremarkable. Insulin levels were increased (33.9mmol/L). Therefore, a diagnosis of hyperinsulinism was made. The patient still suffered from daily hypoglycaemic episodes despite multiple medications including diazoxide, hydrochlorthiazide, nifedipine and octreotide; therefore, further investigations were performed. Genetic studies found ABCC8c.1792C>Tp.Arg598* heterozygous novel mutation and subsequent molecular analysis confirmed paternal in origin. Nuclear imaging with 18FDOPA- PET scan showed increase 18F-DOPA uptake in a focal area of the pancreas suggestive of focal type of congenital hyperinsulinism. In view of the above findings, laparoscopic partial pancreatectomy of the focal lesion was performed; however, post-operatively the patient still required high glucose infusion requirement suggestive of residual disease. A second surgery involving a subtotal pancreatectomy by excising 80% of the pancreas led to significant improvement clinically with occasional episodes of hypoglycaemia. Subsequent histopathology confirmed focal hyperinsulinism with several foci suggestive of multifocal hyperinsulinism.
Discussion
Multifocal hyperinsulinism is poorly described in the literature. It appears to have similar characteristics and genetic mutations as focal type disease [9]. Arnoux described one case of persistent hypoglycaemia immediately after surgery which was believed to be accounted for an exceptional distinct second and third focal form in the same patient as in the case of our patient [5]. Multifocal forms of the disease may be missed on 18F- DOPA-PET scan if the lesions are close by because the resolution of the method may not allow separation of their images; which was observed in three out of 150 cases in a study by Delonlay [9]. This atypical form presents a surgical challenge. Intraoperative identification of the lesion is challenging as no significant macroscopic changes are seen in focal hyperinsulinism; therefore, confirmation of complete resection of the lesion can only be confirmed after the actual resection. In addition, as in the case of multifocal type disease, identification of second and third focal forms may not be suspected even after the complete resection of the focal lesion. This may subject the patient to subsequent surgical procedures. Novel imaging techniques may be necessary to improve the accuracy of focal lesion identification and may be ‘labeling’ of the lesion in order to aid intraoperative identification. Similarly, to the case described by Arnoux our patient still had some but significantly less frequent episodes of hypoglycaemia requiring continued medical treatment even after surgery [5].
Conclusions
Congenital hyperinsulinism is a common cause of persistent hypoglycaemia in infancy, which if not diagnosed and treated promptly may lead to permanent brain injury. The management of congenital hyperinsulinism has changed remarkably over the last decade with the new advances in molecular genetics and imaging studies. In diazoxide resistant patients, the search for focal or diffuse forms of the disease via genetic mutation studies and 18-F-DOPA PET scan is necessary, as many focal forms are cure by partial pancreatectomy without suffering from postoperative diabetes mellitus. However, in multifocal disease, it is often difficult for all lesions to be identified in the current imaging modality and intraoperatively there is no intraoperative imaging that would assist and ensure the resection of all lesions; therefore, they may not be as easily ‘cured’ as in the cases of single focal disease. The development of novel imaging techniques may be necessary to improve the identification and localisation of these focal lesions. In cases of diffuse form of the disease, near-total pancreatectomy may be necessary for disease control; however, it is often only performed in medically non- responsive and severe cases due to the risk of postoperative diabetes mellitus. Despite our progress in molecular genetics, there are still genes yet to be identified. Therefore, further genetic studies looking for new involved genes may lead to novel therapeutic therapies.
References
-
Petraitiene I, Barauskas G, Gulbinas A, Malcius D, Hussain K, et al. (2014) Congenital Hyperinsulinism. Medicina 50(3): 190-195.
-
Stanley CA (2016) Perspective on the Genetics and Diagnosis of Congenital Hyperinsulinism Disorders. J Clin Endocrinol Metab 101(3): 815-826.
-
Yorifuji T (2014) Congenital hyperinsulinism: current status and future perspectives. Annals Ped End Metab 19(2): 57-68.
-
Yorifuji T, Horikawa R, Hasegawa T, Adachi M, Soneda S, et al. (2017) Clinical practice guidelines for congenital hyperinsulinism. Clin Pediatr Endocrinol 26(3): 127-152.
-
Arnoux J, Verkarre V, Saint-Martin C, Montravers F, Brassier A, et al. (2011) Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis 6: 63-77.
-
Rozenkova K, Guemes M, Shah P, Hussain K (2015) The Diagnosis and management of hyperinsulinaemic hypoglycaemia. J Clin Res Pediatr Endocrinol 7(2): 86-97.
-
Kappor R, Flanagan S, James C, Shield J, Ellard S, et al. (2009) Hyperinsuliniaemic hypoglycaemia. Arch Dis Child 94(6): 450-457.
-
Thornton P, Stanley C, Leon D, Harris D, Haymond M, et al. (2015) Recommendations from the Pediatric Endocrine Society for Evaluation and Management of Persistent Hypoglycemia in Neonates, Infants, and Children. J Pediatr 167: 238-245.
-
Delonlay P, Simon A, Galmiche-Rolland L, Giurgea I, Verkarre V, et al. (2007) Neonatal hyperinsulinism: clinicopathologic correlation. Human Pathology 38(3): 387-399.
- Shaping Healthy Futures: Pediatric Endocrine Breakthroughs of 2025
- Precision Medicine in Obesity: Customizing Treatment for 2025
- The Thyroid Revolution: How 2025 is Redefining Hormone Health
- Editorial- Targeting Immunometabolism for Generating Innovative Therapies for Cancer
- Current Knowledge of Chickenpox
- Correlation of Preinjection Values of Gonadotropins and Estradiol Level with Clinical and Radiologic Evidence of Sufficient Pubertal Suppression in Girls with Central Precocious Puberty