An Update on the Risk Factors Correlating NAFLD with Cardiovascular Disease: Specifically Mitochondrial - Fatty Acids β Oxidation in Liver with Therapeutic Approaches of Avoidance of CVD Associated Mortality-A Systematic Review
Nonalcoholic fatty liver disease (NAFLD) is a rapidly escalating disorder which impacts a large population worldwide. Nevertheless, cardiovascular disease (CVD) represents the biggest etiology for mortality with regards to patients of NAFLD. Atherogenic dyslipidemia, possessing the properties of plasma hypetriglyceridemia, enhancement of small dense (low density lipoproteins)LDL’s particles in addition to reduction of high density lipoproteins cholesterol HDL-C) concentrations are generally seen in patients with a presentation of NAFLD. Thus here we conducted a systematic review utilizing search engine PubMed, Google scholar ;web of science; embase; Cochrane review library utilizing the MeSH terms like NAFLD; CVD; atherosclerosis; insulin resistance(IR); NASH; chronic heart failure; dyslipidemia; hepatokines; endothelial impairment; pro inflammatory cytokines; FA’s oxidation; SREBP1c; ChREBP; Sirtuin; LKB1; lipogenesis; mitochondrial lipid β oxidation; genetic mutations from 2010 to 2022. We found a total of 500 articles out of which we selected 143 articles for this review. No meta-analysis was done. Thus here we have detailed the more recent genetic corroboration, with provision of distinctive type of metabolic pathways implicated in NAFLD pathogenesis. Assessment of the genetic results that are accessible pointed that the crucial process that correlated NAFLD modulated dyslipidemia, along with escalation of risk of CVD implied is the changes in the handling of fatty acids β oxidation in the liver mitochondria. NAFLD correlated genes possessing reported anti-atherosclerotic or cardio protective actions in addition to existent Pharmacologic approaches that are concentrated on tackling both treatment of NAFLD in addition to reducing the risk of CVD. Further research demonstrates that inhibitors of de novo’’ lipogenesis (DNL) might prove to be of benefit.
Introduction
Nonalcoholic fatty liver disease (NAFLD) prevalence has been enhancing at a rapid pace, influences 25-45% of the adult population globally besides up till 70% in type2 Diabetes mellitus (T2DM) besides obesity patients group [1]. An association of NAFLD exists with numerous co- morbidities (hypertension, obesity, Metabolic Syndrome (MetS) hyperlipidemia). Nonalcoholic steatohepapititis (NASH), represents a greater robust type implicated in 2-7% of adult besides propagating to cirrhosis &Hepatocellular carcinoma (HCC) subsequently. Cardiovascular disease (CVD) is a usual etiology of mortality amongst NAFLD patients [2]. It is considerably significant, that despite all NAFLD kinds correlated with CVD, maximum probability is existent with NASH besides advancements of fibrosis [3].
The precise mode for correlation amongst NAFLD with CVD has not been corroborated. Recently, variable stimulators suggested, accounting for propagation of NAFLD along with exaggeration of atherogenesis, dyslipidemia, chronic inflammation besides endothelial impairment [4]. The Adipose tissue (AT) expansion that accompanies causes an initiation of the pro inflammatory cascade along with nuclear factor κB (NFκB) as well as c-jun-N-terminal kinase (JNK) pathways. Moreover the complications might be inclusive of insulin resistance (IR), hepatic system wide level escalation of generation of cytokines as well as chemokines (like Tumor necrosis factor alpha (TNFα) as well as interleukin-6 (IL-6), C Reactive Protein (CRP) along with others, formation of procoagulant factors (factor VIII, endothelin, transforming growth factor beta (TGF-β), fibrinogen etc.) in addition to hepatokines impairment of glucose along with lipid metabolism [5].
Generation of neointimal plaques associated with atherosclerosis in large arteries result in stimulation of cardiovascular (CV) processes (like stroke, myocardial infarction (MI). Atherosclerosis represents a chronic disease that lasts for decades at the time of which inflammation, calcification, fibrosis along with lipid getting deposited results in alteration in the constituents of plaques associated with atherosclerosis [6]. Utilization of variation in methodologies have been attempted for evaluating the determination of plaque characteristics besides assessment of risk of CVD; utilization of invasive techniques is conducted usually where advancement of stage has occurred (angiography, optical coherence tomography or intravascular ultrasonography) in addition to noninvasive is commonly utilized for the primary diagnosis (positron emission along with computer tomography, determination of carotid intima-media thickness, etc.,) [7].
Outcomes of numerous cross -sectional studies, systematic reviews and meta-analysis pointed to NAFLD resulting in escalation of risk of atherosclerosis along with facilitation of generation of unstable plaques [8]. Additionally, genetic proof pointed to NAFLD manipulated dyslipidemia is the key factor for the enhancement of risk of CVD [9]. Whereas numerous genetic polymorphism areas along with mutations possess the correlation with CVD[10] as well as NAFLD [11], certain NAFLD resulting in facilitation of Single nucleotide polymorphisms (SNP) have been detailed as resulting in reduction of risk of CVD [12, 13]. Other workers nevertheless did not observe such protection [14]. Having reviewed the various probable etiologies of NAFLD/NASH along with potential therapies [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26] here we decided to update on the association of NAFLD along with generation of heart diseases in NAFLD/NASH we decided to carry out a systematic review.
Methods
Here we conducted a systematic review utilizing search engine pubmed, google scholar; web of science; embase; Cochrane review library utilizing the MeSH terms like NAFLD; CVD; atherosclerosis; insulin resistance (IR); NASH; chronic heart failure; dyslipidemia; hepatokines; endothelial impairment; proinflammatory cytokines; FA’s oxidation ; SREBP1c; ChREBP; Sirtuin; LKB1;lipogenesis; mitochondrial lipid β oxidation; genetic mutations from 2010 to 2022.
Results
We found a total of 500 articles out of which we selected 143 articles for this review. No meta-analysis were done.
Correlation amongst Liver & Heart Diseases
Different modes reasoning out intricate association amongst CVD with NAFLD got pointed out. The key part of insulin resistance (IR) was appreciated regarding crucial role in etiopathogenesis of NAFLD & NASH [27]. Additionally, IR influences numerous physiological events causing hyperglycemia, dyslipidemia resulting in low grade chronic inflammation activation, ectopic lipid accrual, Oxidative stress (OS) besides endothelial impairment [28]. Escalated serum ferritin, major protein implicated in iron storage is commonly observed in NAFLD patients besides correlated with IR [29]. Together these processes produced a CVD facilitating proatherogenic milieu [30]. Together with changed immune cells population in NASH patients [31] pointed to an immune besides chronic proinflammatory correlation amongst IR, CVD with DM [32]. Fetuin A, a glycoprotein liberated from AT besides liver stimulated proinflammatory cytokine generated from adipocytes besides macrophages besides acting as biomarker for variable chronic inflammatory diseases. Fetuin A (i.e., fatty acid (FA) carrier) is further escalated in NAFLD/ NASH patients [33]. Recently, it was illustrated that Fetuin A hampered insulin receptor tyrosine kinase in muscle besides liver resulting in IR [34]. The quantity of Fetuin A was associated with hypetriglyceridemia, not significantly correlation with ischemic stroke besides other CVD was observed [35], whereas other researchers, pointed it a key factor regarding chronic heart failure (HF) diagnosis [36].
Variable dyslipidemia quantities generally are presenting in patients of NAFLD/ NASH (reduced high density lipoprotein (HDL) besides enhanced of low DL (LDL), particles, triglycerides (TG) besides acting as noninvasive markers for NAFLD diagnosis [37]. Nevertheless this kind of lipid profile is considered atherogenic besides associated with robustness of cardio metabolic risk [38]. Liver serves as the central organ accountable for metabolism amongst which TG, cholesterol (TC) are the ones of maximum significance. Whereas low HDL-C has been considered as a well appreciated biomarker for NAFLD besides a risk factor for CVD the precise mode for this correlation is getting evaluated [39]. The most appropriate HDL function is it facilitates reverse cholesterol transport (RCT) aiding extra cholesterol deletion from macrophages with> excretion from body via bile. Interest evoked in RCT as attractive therapeutic target for CVD risk reduction [40]. Nevertheless, therapeutic enhancement of HDL-C quantities with drugs utilization not found to be advantageous for the reducing CVD risk pointing to a greater complicated association amongst, HDL-C besides CVD [41].
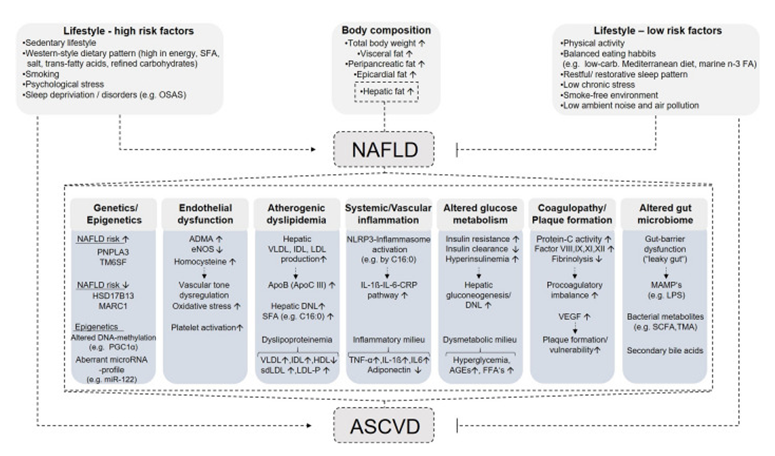
Figure 1: Courtesy ref no-143-Pathophysiological mechanisms linking NAFLD and CVD. Saturated fatty acids, OSAS obstructive sleep apnea syndrome. carbohydrates, n-3 FA omega-3 fatty acids, NAFLD non-alcoholic fatty liver disease; PNPLA3 patatin-like phospholipase domain-containing protein 3, TM6SF transmembrane 6 superfamily 2 human gene, HSD17B13 hydroxysteroid dehydrogenase 17beta 13, MARC1 mitochondrial amidoxime reducing component 1, PGC1α peroxisome proliferator-activated receptor gamma coactivator 1-alpha, miR micro-RNA, ADMA asymmetric dimethylarginine, eNOS endothelial nitric oxide synthase, VLDL very-low-density lipoprotein, IDL intermediate-density lipoprotein, LDL low-density lipoprotein, ApoB apolipoprotein B, ApoC III apolipoprotein C3, DNL de novo lipogenesis, HDL-C high-density lipoprotein cholesterol, sdLDL small dense low-density lipoprotein, LDL-P low-density lipoprotein particles, C16:0 palmitic acid, IL-1β interleukin-1β, IL-6 interleukin-6, CRP C-reactive protein, AGEs advanced glycation end products, FFAs free fatty acids, VEGF vascular endothelial growth factor, MAMPs microbe-associated molecular pattern, LPS lipopolysaccharide, SCFA short-chain fatty acids, TMA trimethylamine; ASCVD atherosclerotic cardiovascular disease. Down arrows (↓) indicate decreased levels, and up arrows (↑) indicate increased levels.
Enhanced serum homocysteine quantities well appreciated as etiology of hepatic Oxidative stress (OS) besides hepatic steatosis [42], associating with liver impairment in NAFLD/ NASH patients [43]. Akin to that serum homocysteine is an independent factor regarding CVD [44]. Activation of toll like receptor 4 (TLR4), by homocysteine is well appreciated causing impaired Ca2+,, nitric oxide (NO), escalated Reactive oxygen species (ROS) generation besides inducing platelet- activation besides endothelial impairment ultimately resulting in CVD [45].
Other association amongst NAFLD with cardiovascular action is proinflammatory cytokine, liberation by liver resulting in system inflammation besides facilitating CVD. The major inflammation induced triggers causing CVD are enhanced plaques generation, changed vascular tone, coagulation besides endothelial function [46]. Variable cytokines (IL-1, IL-6 C Reactive Protein [CRP besides TNFα 6) ]) concentrations considered systemic inflammation markers are enhanced in NAFLD patients [47]. Recently liver steatosis, fibrosis correlation pointed with diastolic heart impairment besides myocardial glucose uptake dysfunction [48]. Additionally hepatic fat concentrations were associated with escalated left ventricular filling pressure occurring prior to HF [49].
Future researcher’s concentrations on early pick up of metabolic markers in liver besides heart effectiveness need optimization prior to appearance of functional besides structural aberrations. Hence in subjects with NAFLD diagnosis feasibility of complications avoidance within the CVS. Here we concentrate on liver lipid homeostasis part, mitochondrial-β oxidation in correlation with NAFLD besides CVD correlated with genetic controlling besides targeted treatments (Figure 1).
Liver-the Major Central Organ for Lipid Metabolism
Liver possesses the key part in glucose, lipid metabolism besides homeostasis. On continuous stress secondary to dysfunctional FA metabolism, considerable lipid amounts accrual result in generation of NAFLD. The major characteristic of NAFLD is hepatic TG accrual that could have resulted by internal (dysfunctional Fatty Acid oxidation [FAO], very low density lipoprotein [VLDL] generation & export), external (some genetic background besides environmental situations) or behavioral (increased FA from diet, circulation, or AT, absence of exercise/physical action) triggers [50]. Loss of balance amongst imported vs. exported generated vs. processed FA’s caused accrual of hepatic lipid, hepatosteatosis besides IR, resulting in hepatic “de novo” lip genesis completing this vicious cycle [51]. Whereas Clarification regarding the precise molecular mode under strict evaluation dysfunctional FA’s metabolism besides ROS generation resulting in chronic inflammation besides impaired mitochondrial function pointed as crucial processes in generating this disease[52].
Homeostasis of Lipids in Liver Fatty Acids (FA’s) β- Oxidation
Carbohydrates along with Fatty Acids(FA’s)are the providers of energy for cells with their uptake from extracellular space along with intracellular liberation is tightly regulated by variable hormones like glucagon, insulin, noradrenaline etc., FA’s esterification, metabolism to lipids 2nd messengers (like ceramide, sphigosine, phosphatidyl inositol b is phosphate, etc.,) or transportation to mitochondria for β- oxidation. Nevertheless, a very long chain fatty acid (VLCFAs) (FA’s possessing over22 as well as greater carbons) activation occurs, that can’t get metabolized in mitochondria, thus need for transportation to peroxisomes [53]. Subsequent to activation of long chain fatty acids(LCFAs)by CoA, transportation of LCFA CoA, ester occurs to mitochondrial matrix through the carnitinepalmoyl transferase (CPT) comprising of 3 proteins, CPT1, CPT2 as well as CACT(acyl carnitine translocase) [54, 55, 56, 57] in Figures 2 & 3 for normal metabolism.
Hampering of CPT1 is feasible by malonyl CoA, obtained from glucose metabolism; hence that makes CPT1, a rate limiting enzyme or step with regards to mitochondrial FAO. Three isoforms of CPT1are existent, (A) That are organ particular for liver, (B) Muscle as well as heart and (C) Brain. Furthermore, over 2 enzymes are implicated in metabolism of malonyl CoA, namely acetyl CoA carboxylase (ACC). ACC can account for the generation of malonyl CoA, along with malonyl CoA decarboxylase (MCD), that is responsible for breakdown of malonyl CoA [58].
Two major isoforms of ACC are present, i.e., ACC 1 as well as ACC 2 possessing variable tissue expression patterns along with functions. ACC 1 is existent in the cytoplasm of all cells, however in lipogenic tissue like AT it is present in abundant quantities [59]. ACC 2 resides in the mitochondria, besides being abundant in oxidative tissue (like skeletal muscle as well as heart) [60]. Hence variable tissues possess a particular ACC 1 /ACC 2 ratio that are a requirement for sustenance of balance amongst, FA’s oxidation along with its generation. Both ACC 1 as well as ACC 2 are present as well as have high expression in liver where both these FA’s oxidation along with its generation occurs considerably. Nevertheless, such a variation of where ACC resides along with its function aids in provision of a chance of development of a pharmacological drug for particular hampering of FA’s
generation along with stimulation of fatty acids oxidation that might be of advantage for certain morbidities like obesity, type2 Diabetes mellitus(T2DM), NAFLD as well as others [61].
5’ AMP-activated protein kinase (AMPK) represents one of the major controllers of this pathway, its action is via phosphorylation along with hampering of ACC, hence result in reduction of expression of FA synthase along with provision of intermediates for the FA anabolic pathway. In the form of a secondary, in addition to long term action, AMPK results in phosphorylation of sterol regulatory element binding protein 1c(SREBP1c), as well as carbohydrate response element binding protein (ChREBP), therefore hampering of the transcription of following lipogenesis genes [62]. Sirtuin proteins (SIRT1 SIRT3) result in stimulation of AMPK through deacetylation of the upstream activator liver kinaseB1 (LKB1) [63]. These days the remarkable significance of Sirtuins has been appreciated in the form of key controllers of lipid metabolism with provision of particular FAO facilitating actions (in skeletal muscle as well as liver) lipolysis (in AT), mitochondrial respiration, brown adipose tissue (BAT) along with food consumption (in the hypothalamus) [64].
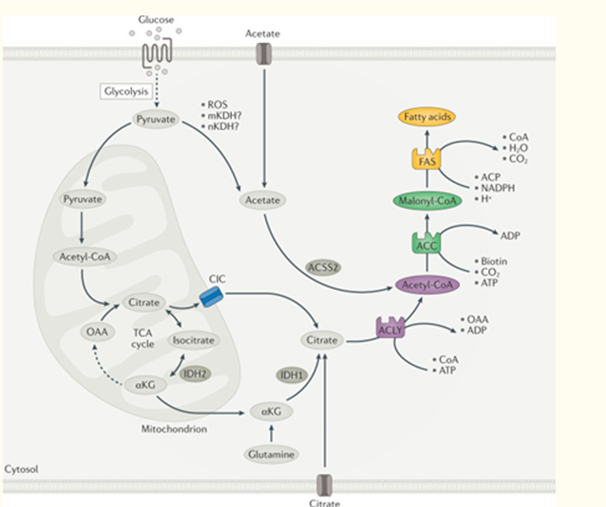
Figure 2: Overview of DNL. Courtesy ref no-142-A series of coordinated enzymatic reactions takes place during fatty acid biosynthesis. Typically, pyruvate produced by glycolysis is converted in the mitochondrion into acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle to produce citrate. In conditions of carbohydrate excess, citrate is exported to the cytosol by the citrate/isocitrate carrier (CIC) and is broken down to acetyl-CoA and oxaloacetate (OAA) by ATP-citrate lyase (ACLY). Acetyl- CoA is subsequently carboxylated by acetyl-CoA carboxylase (ACC) to generate malonyl-CoA, which is considered the first committed metabolic intermediate in fatty acid synthesis. Utilizing seven malonyl-CoA molecules and one acetyl-CoA primer, the synthesis of palmitate (16:0 fatty acids) is completed by repeating a cycle of condensation, reduction, condensation and dehydration catalysed by fatty acid synthase (FAS). An alternative carbon source of de novo lipogenesis (DNL) is acetate, which can be produced de novo from glucose through non-enzymatic and enzymatic reactions. Acetyl-CoA synthetase 2 (ACSS2) catalyses the reaction of acetate and CoA to form acetyl-CoA, which is subsequently used for fatty acid biosynthesis. With hypoxia or CIC deficiency another alternative pathway for DNL is reductive carboxylation of glutamine via cytosolic isocitrate dehydrogenase 1 (IDH1) and mitochondrial IDH2. αKG, α-ketoglutarate; ACP, acyl carrier protein; mKDH, mitochondrial ketoacid dehydrogenase; nKDH, nuclear ketoacid dehydrogenase; ROS, reactive oxygen species.
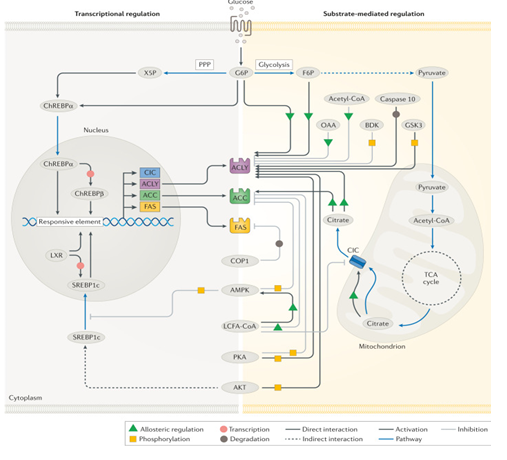
Figure 3: Physiological regulation of DNL. Courtesy ref no-142-Regulatory mechanisms of de novo lipogenesis (DNL) involve allosteric regulation, covalent modifications and transcriptional changes. Allosteric activators include citrate, glucose 6-phosphate (G6P) and fructose 6-phosphate (F6P) while oxaloacetate (OAA) and long-chain fatty acyl (LCFA)- CoAs are allosteric inhibitors. Regulatory phosphorylation is facilitated by several enzymes including AMP-activated protein kinase (AMPK), AKT, branched-chain α-keto dehydrogenase kinase (BDK), glycogen synthase kinase 3 (GSK3) and protein kinase A (PKA), whereas caspase 10 and constitutive photomorphogenic 1 (COP1) facilitate the degradation of ATP-citrate lyase (ACLY) and fatty acid synthase (FAS), respectively. Transcriptional modifications are regulated by two major transcription factors, sterol regulatory element-binding protein 1c (SREBP1c) and carbohydrate-responsive element-binding protein (ChREBP). Additional transcription factors, such as liver X receptor (LXR) are also implicated in the transcriptional regulation to varying degrees of importance depending on the cell type. ACC, acetyl-CoA carboxylase; CIC, citrate/isocitrate carrier; FAS, fatty acid synthase; PPP, pentose phosphate pathway; TCA, tricarboxylic acid; X5P, xylulose 5-phosphate.
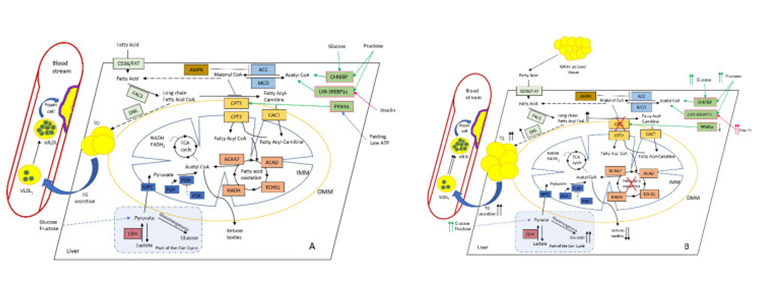
Figure 4: Courtesy ref no-55-Altered hepatic lipid metabolism in normal (A) and NAFLD; (B) conditions. Increased diet supply of glucose and fructose affects ChREBP (carbohydrate-responsive element-binding protein), SREBP1c (sterol regulatory element-binding protein-1c), and LXR (liver X receptor) TFs, which stimulate malonyl CoA synthesis. PPARα (peroxisome proliferator-activated receptor alpha) normally activates CPT1 under fasting and low-ATP conditions. As a key intermediate, malonyl CoA inhibits CPT1, thus reducing FAO. This leads to the accumulation of long-chain fatty-acid CoA (which could also be delivered from surplus adipose tissue), and stimulates DNL with the subsequent rise in intrahepatic TG and plasma TG levels, further increasing large VLDL1 and the formation of small dense LDL, which favours foam cell formation and ultimately atherosclerosis. During the progression of NAFLD, the production of ketone bodies progressively reduces while hepatic glucose synthesis and output increases, thus further promoting IR and the rise in insulin level. Colour coding as follows: PPARα and other TFs are depicted in pale green; primarily FA-metabolising enzymes (FAT (fatty-acid translocase) and FACS (fatty-acid synthase)) and DNL (de novo lipogenesis) are highlighted in pale blue; malonyl-CoA-metabolising enzymes ACC (acetyl-CoA carboxylase) and MCD (malonyl-CoA decarboxylase) are highlighted in blue; CPT system enzymes (CPT1, CPT2, and CACT) are depicted in light brown; AMPK (AMP-activated protein kinase), the main regulator of CPT system are depicted in brown; FAO enzymes ACAA2 (acetyl-CoA acyltransferase 2), ACAD (acyl-CoA dehydrogenase), ECHS1 (enoyl-CoA hydratase, short chain 1), and HADH (hydroxyacyl-CoA dehydrogenase) are depicted in in pale red; pyruvate metabolism enzymes MPC (mitochondrial pyruvate carrier 1), PDP (pyruvate dehydrogenase phosphatase), PDH (pyruvate dehydrogenase), and PDK (pyruvate dehydrogenase kinase) are depicted in dark blue; and LDH (lactate dehydrogenase) is depicted in red. Abbreviations: IMM and OMM, inner and outer mitochondrial membrane, respectively.
Mitochondrial FAO in the liver result in full oxidation to CO2 or partly when ketone bodies, that represents an exported kind of energy possessing molecules get generated. The results in the context of CPT1A amounts of expression along with action, besides rate of mitochondrial FAO are debatable with considerable, dependence on the utilization of model system, amounts of free fatty acids (FFA) in addition to other experimental situations [65]. For giving the reasons of these variations of modes have been pointed)the amounts of malonyl CoA are key that are dependent on the ratio of ACC/MCD proteins ii) the physical characteristics of the mitochondrial membrane might alter the sensitivity of CPT1A to malonyl CoA [66] along with iii)the accessible pool of FFAs as well as other lipid intermediates that could result in activation of various transcription factors implicated in denovo FAs generation, uptake, transportation in addition to oxidation[67].
The present model gives the explanation for the correlation amongst FFAs as well as FAO in the form of hormetic action (a dose-response phenomenon characterized by low-dose stimulation and high-dose inhibition) when a mild or vanishing enhancement in accessible FFAs results in an advantageous escalation of FAO with greater energy formation. Nevertheless, continuous along with significant FFAs liberation result in an escalation of electron flux in electron transport chain (ETC), over generation of ROS along with generation of toxic aldehydes that cause injury to mitochondrial protein, lipids as well as -5DNA result in morphological along with functional disruptions [68]. In the form of a probable approach for avoidance, of this type of adverse actions the liver possesses the capacity of shifting the balance from total FA oxidation towards generation of ketone bodies [69]. Nevertheless, akin to the FAO pathway this approach possesses its restrictions apart from result in certain complications at the time of continuous FFAs liberation along with reduction in energy expenditure [70].
Crosstalk along with Glucose Metabolism
The present diet (in particular in classical western diets [WD]) possess a greater amount of simple fructose in addition to glucose fructose dependent saccharides that constitute as risk factors for the generation of different metabolic complications, like obesity,T2DM, NAFLD,CVD as well as others [71]. Linolenic acid represents, a) polyunsaturated omega 6 fatty acids (PUFA’s), is further broadly present In WD is correlated with obesity, IR, along with CVD [72]. We possess the knowledge that fructose acts as a substrate for the generation, of FA’s along with stimulation of the transcription factor for de novo lipogenesis along with formation of triglycerides (TG), SREBP1c, as well as ChREBP [73]. Concomitantly, fructose results in reduction of FAO through 2 major modes; namely:
- Escalation of amounts of hepatic malonyl CoA.
- Along with direct changing the expression of hepatic genes that account for lipid accrual as well as elimination [74].
The distinctive part of fructose effect is a temporary reduction in the intracellular amounts of phosphate along with ATP that possesses a correlation with the uric acid formation along with turnover of nucleotide. A reduction in the ATP amounts results in induction of a stepwise reaction, that is inclusive of the induction of Oxidative stress (OS), a temporary blockade of protein generation in addition to mitochondrial impairment, that possesses the crucial part in modulation of fructose actions [75]. In view of variable CPT1 isoforms possessing various sensitivities to malonyl CoA, where liver isoforms possesses30-100 fold lesser sensitivity in contrast to muscle as well as heart isoforms [76], fructose’s malonyl CoA independent action on the liver mitochondrial action might possess greater prominent along with result in facilitation of greater level of mitochondrial impairment. In addition diet obtained nutrients might control mitochondrial function through post-translational modifications (malonylation, acetylation, succinylation, as well as others) [77]. Nevertheless, we have already detailed these earlier in DM nephropathy [78].
Part of Perilipin in NAFLD as well as Atherosclerosis
Plin5 (Perilipin 5) represents a significant member of the Perilipin protein, that is present in mammoth amounts in tissue with presentation of active lipid catabolism like heart, skeletal muscle, BAT as well as liver [79]. NAFLD has the properties of escalation of accrual of lipid droplet (LDs) in the liver, along with escalation of expression of PLIN5 [80]. Plin 5 is believed to be the major LDs developing along with coating protein taking into account the restoration of hepatic TG’s in LDs as well as hampering lipolysis. Thus it is not astonishing that over expression of Plin5 results in deterioration of hepatosteatos [81], in addition to blockade of stellar cell activation [82], however without deleterious actions on IR. Conversely deficiency of PLIN5 results in impairment of insulin signaling transduction along with the generation of IR [83].
More recently, studies have pointed out the molecular modes offering an explanation for these findings. The C terminal part of Plin5 (ie443-63aa) enrolls the mitochondrial for connecting the LDs. This, LD - mitochondrial connection is needed for appropriate provision of FA’s to mitochondria, generation of lipids in addition to lipid expansion [84]. These characteristics pointed to the part of mitochondria in the development of TAG’s along with phospholipids in view of enzymes located in outer membrane of mitochondria glycerol-3 phosphate acetyl transferase I as well as 2(GPAT) along with 1-Acyl glycerol-3 phosphate acetyl transferase (AG-PAT) are implicated, in the bio generation of lysophosphatidic acid along with phosphatidic acid, respectively [85]. Mitochondrias that are correlated with LDs possess the escalation of capacity for pyruvate oxidation, electron transport as well as generation of ATP, a reduction in ability for β oxidation as well as distinctively lesser fusion -fusion dynamics [86]. Furthermore, Plin5 was illustrated to restrict FA’s toxicity, clearance of deleterious proteins from the outer membrane of mitochondria as well as confer protection against OS [87].
PLIN5 was implicated, in the inflammatory reaction via the activation, of the NLRP3 (NLR family pyrin domain containing 3) inflammasome, thus correlating with NAFLD/ NASH propagation [88]. Additionally, a greater amount of Plin5 was the observation in Hepatocellular carcinoma (HCC) biopsy specimens [89]. Nevertheless, the results with regards to the part of PLIN5 in the generation of HCC as well as metastasis is restricted till date with requirement of further evaluation of the precise part of PLIN5 in HCC.
Intriguingly deficiency of PLIN5 was correlating with the propagation of atherosclerosis, in addition to the way it was illustrated in double knockout mice (Apo E-/- , Plin5-/-) which generated atherogenesis of greater robustness(associated with escalation of TG,TC, along with LDL-C amounts with reduction in HDL-C amounts) in addition to exaggerated inflammation, apoptosis, lipid accrual as well as OS. Mutant mice resulted in facilitation of propagation of atherogenesis over the full aorta, aortic arch in addition to full abdominal aorta area [90]. Furthermore, PLIN5 was implicated, in thermal control as well as adapting to cold stress. In a study in which mitochondria along with LDs Identification was feasible from the liver of mice which had been housed in chronic cold stress documented that mitochondrial TCA cycle as well as retinol metabolism were escalated whereas there was no impact on Oxidative phosphorylation. Adaptation of liver to cold stress situations implicated escalation of Plin5 along with major urinary proteins (MUPs), while there was an astonishing reduction in MPC [91]. Previously the part of thermogenesis in the generation of metabolic diseases was correlated mainly with AT (inter action along with transition amongst WAT as well as BAT) [92]. Nevertheless, the detailed cold- adaptation associated part of Plin5 further pointed to its part with regards to homeostasis along with of liver lipids in the generation of NAFLD [91].
These observations established that PLIN5 is a key pleiotropic controller of hepatic lipid metabolism, thermogenesis along with, inflammatory reaction implicated in NAFLD/NASH along with generation of atherosclerosis in addition to propagation. Hence PLIN5 is an attractive therapeutic target for NAFLD as well as atherosclerosis, besides probably for certain other metabolic diseases [93].
Part of Liver Mitochondria in the Generation of CVD Facilitating Dyslipidemia
Some bit of atherogenic dyslipidemia is existent in patients of NAFLD/NASH which possess the properties of TC/ HDL-C, LDL-/HDL-C TG/HDL- Cratios [94]. It is well believed that liver possesses a central part in the development/ clearing of all lipoproteins (HDL) along with Apo lipoproteins (ApoB48 as well as ApoB100)for establishment of a robust connection amongst NAFLD/NASH correlated with metabolic impairment along with escalation of CVD risk [95]. Here our concentration is on the recent insights with regards to association amongst genes understood to confer protection/ result in NAFLD abrogate/ deteriorate the phenotype of NAFLD, change liver particular lipid accrual, or have an impact on the lipid profile at a system wide level .
Numerous genes are known to be responsible for FAO conditions that are not detailed here in view of lack of NAFLD/ NASH correlated phenotype, nevertheless, these genes are of importance [96]. Assessment of mutations detailed pointed to the existence of central part of liver mitochondrial FAO in association amongst NAFLD along with CVD risk .The crucial points being:
- Escalation of FAO might result in amelioration of NAFLD symptomatology however lead to hypertriglyceridemia as well as OS injury to the liver.
- Appropriate provision of antioxidant might support FAO by affecting neutralization of escalated ROS total lack of FAO in liver mitochondria result in resistance to HFD obesity along with NAFLD in addition to serum dyslipidemia as well as hepatic OS.
- Appropriate mitochondrial turnover might support efficacy of FAO along with mitigate NAFLD symptoms.
- Total lack /decrease of FAO resulted in enhancement of energy expenditure (EE) at the system wide manner in addition to suppression of adiposity.
- Once FAO has been rendered ineffective other modes of stimulation of oxidation in other chambers/cells (peroxisomes as well as microsomes, macrophages besides others).
Hence evaluation of mutations accumulated corroborated the posit of hormetic action [96], when the disappearing escalation of FFA input was of advantage along with resulted in stimulation of FAO, provision of greater energy output. Enhancement or protracted stimulation of FAO is deleterious; the pool of antioxidants which have been deleted does not possess the capacity of tackling the escalation of generation of the ROS that results in mitochondrial along with liver injury besides dyslipidemia causing further complications associated with the CVS. Different probable pathways take into account these actions. Over generation of ROS might result in lipid peroxidation resulting in development of 4hydroxynonenal -CPT1 adduct, implicated in dysfunctional FAO, in addition to elimination of lipids from hepatocytes. Additionally ROS act on PUFA resulting in lipid peroxidation along with generation of toxic aldehydes by products (hydroxyl 2nonenal along with malondialdehyde (MDA) that possesses greater half-life in contrast ROS as well as can be transported from the area of generation towards distant intracellular as well as extracellular targets thus result in accelerated actions of OS [97].
During normal physiological situations oxidation of long as well as medium chain FA’s gets carried out basically by mitochondrial β - oxidation system, with limited assistance from the peroxisomal system .VLCFA’s do not work as substrates for CPT1 hence do not possess the capacity of gaining entry into the mitochondria, nevertheless preferential VLFA’s actions are seen in the form of substrates for peroxisomal β-oxidation [98]. Inspite of the existence of total enzymatic machinery with regards to peroxisomal β- oxidation under normal circumstances is incomplete with the shuttling towards mitochondria of the final products of peroxisomal β- oxidation with the idea of achieving finishing of total oxidation to CO2, H2O along with ultimate energy development. Furthermore, utilization of certain products of peroxisomal β- oxidation might occur in other metabolic pathways (like taking part in the generation of taurine as well as glycine conjugates that gets followed by export to the biliary duct)[99].
FA’s further might undergo ω oxidation via the effect of a microsomal oxidase where utilization of molecular oxygen in addition to both alcohol as well as aldehyde dehydrogenase for the generation of dicarboxlixc acids. Further break down of dicarboxlixc acids might take place by peroxisomal β- oxidation to succinate as well as acetyl CoA, or total oxidation following transport into the mitochondrial β- oxidation system [100]. At the time of normal physiological situations ω oxidation constitutes a minor pathway of FA metabolism however in case of failure of mitochondrial β- oxidation this pathway can assume significance with escalation of ω oxidation action with generation of escalated dicarboxylic acids which are non-specific particular markers of aberrations in mitochondrial FAO [101, 102].
If total absence of mitochondrial FAO the escalation of the amounts of serum hepatokines were found like insulin like growth factor binding protein1 (IGFBP1), Growth Differentiation factors15, as well as Fibroblast growth factor 21 (FGF21) which pointed to their with regards to physiological adjustment to the greater lipid load from a high fat diet (HFD) [103]. Compensation is tried by the liver for the absence of FAO by up regulation of Oxidative programming along with looking for escalation of catabolism in peripheral tissues with the aid of liberated hepatokine [104]. The basic target of these hepatokines is escalation of energy expenditure from BAT in addition to result in browning (for white) [105]. Moreover macrophages are further implicated in participation in BAT thermogenesis [106]. The significance of part of macrophages was further corroborate by researchers recently where macrophage FAO by definition was that they conferred protection against atherosclerosis, whereas hampering of macrophage FAO might cause escalation of foam cell generation as well as hence result in acceleration of atherosclerosis [107]. Intriguingly hampering of FAO resulted in escalation of ROS amounts possibly secondary to collection of toxic partly metabolized FAO substrates [108]. In this context NAFLD correlated genes revealed anti-atherosclerotic or confer protection to heart, thus further here we outline certain cases where we possess knowledge with regards to metabolic along with genetic NAFLD.
Protection Conferred to Heart
General control no depressible 2 (GCN2) represents a sensor of availability of amino acids which occur in numerous organisms. In case of mammals the maximum amounts of expression of GCN2 was estimated in liver along with brain. Consumption of restricted proteins in diet result in activation of GCN2 thus resulting in phosphorylation of eukaryotic initiation factor2 alpha (eIF2) for hampering translation of global protein along with stimulation of de novo’’ amino acids bio generation for restoration of homeostasis [109]. Furthermore downstream targets are activating transcription factor4 (ATF4) along with C/EBP homologous protein (CHOP) that results in up regulation of autophagy in addition to pathways for bio generation [110]. Moreover, the participation of GCN2 was demonstrated in the control of hepatic lipid metabolism. Like the deficiency of GCN2, resulted in significant amelioration of HFD induced liver impairment, hepatic steatosis as well as IR through controlling of lipogenic genes (like SREBP1/ PPAR-γ) in addition to their downstream targets (fatty acid synthase [FASN], CD36, SCD1) [111]. Concentrations of rest of researchers has been on the combination of action of deficiency of GCN2 along with exercise on hepatic steatosis in addition to implication of AMPK1/SIRT1/PPARα pathway [112].
Intriguingly deficiency of GCN2 was further illustrated to possess the action of conferring protection to heart in case of hearts of DM patients. Specifically significant of OS, cell demise along with lipid accrual in case of GCN2 knockout through hampering eIF2/ATF4/CHOP signaling with subsequent, decrease in Bcl2/Bax ratio in addition to uncoupling protein 2(UCP2) expression [113]. Hence targeting GCN2 for treatment might work out to be an attractive approach with regards to diabetic cardiomyopathy along with in the form of therapy for reduction of the cardiotoxic deleterious actions of the commonly employed anti-cancer drugs like doxorubicin.
More recently, the work of researchers aided in illustrating the mode of the risk of cardiovascular disease in sub kinds of NAFLD (metabolic along with genetic). It has been established that carriers of numerous SNP areas along with mitochondrial mutations possess greater proneness to NAFLD [11]. Nevertheless, conferring protection against coronary artery disease (CAD) was illustrated for different SNP’s of this type [12], which pointed that each mutation region might implicate a distinctive mode of NAFLD proneness/with CVD avoidance [13]. Actually recent meta- analysis research in the context of NAFLD proneness genes do not result in CAD intrinsically [13]. Of the carriers of NAFLD SNP’s, a robust, association visualized total cholesterol (TC), along with, LDL-C with CAD, however, not for plasma triglycerides (TG), along with HDL-C. Enhancement of outflow of FAs as well as de novo’’ lipogenesis start accrual of fat in the liver besides push for more generation of VLDL which tilts the plasma lipid balance towards a proatherogenic along with CVD facilitating miieu [39]. Nevertheless, certain NAFLD correlated SNP’s result in dysfunctional VLDL liberation (TM6F2 as well as PNPLA3 [Palatin –like PhosphoLipaseA2Domain -Containing3] [114], as well as microsomal triacylglycerol transfer protein [MTTP], along with PEMT) hence resulting in reduction of plasma lipids, with hence provision of protection conferred to heart [115]. At present PNPLA31148M has been the most extensively evaluated NAFLD correlated mutation [116]. Solely the Pnpla3148M variant has not been the observation of being deleterious along with experimental mice kept on a standard diet possessed a normal amounts of liver fat [117]. Nevertheless, the amounts of liver fat were 2-3 fold greater on consumption of a high sucrose diet with about 40 fold greater amounts of PNPLA3 protein on hepatic LDs whereas no alterations in Pnpla3 mRNA [117]. Furthermore it was illustrated that a mutant variant possessed the capacity of avoiding ubiquitination, in addition to proteasomal break down that resulted in significant accrual of protein on LDs [118]. Under normal physiological conditions, PNPLA3 works as a PUFA particular lipase or transacylase that resulted in PUFA possessing phosphatidyl choline (PC) or diacyl glycerols (DAG) whose utilization, is feasible for generation of PC [119]. Noticeably, Pnpla33148 variant along with deficiency of PNPLA3 possessed akin actions on liver lipid metabolism, hence resulting in avoidance of mobilizing TG in addition to resulted in lipid accrual in hepatocytes. In case of some situations it possessed the capacity of resulting in generation of NAFLD; nevertheless, for the CVS this TG getting retained in the liver caused a positive system wide action on the serum lipid profile resulting in reduction in the risk of CVD [120].
Transmembrane6 super family member 2 (TM6SF2) represents a transmembrane protein that resides in the endoplasmic reticulum (ER) along with Golgi of enterocytes as well as hepatocytes. Significant reduction of TM6SF2 protein occurs in case of E167K mutations which result in hepatic accrual of TG however lesser plasma amounts of LDL-C hence account for proneness for NAFLD along with protection from CVD respectively [115]. Deficiency of TM6SF simulates the NAFLD phenotype whereas liver particular TM6SF2 resulted in escalation of plasma TC along with LDL-C amounts [121]. ER lipid raft protein1 as well as 2(ERLIN) proteins represents ER residing trans membrane glycoprotein which take part in the control of cholesterol bio generation pathway by blockade of export of SREBPs from the ER to Golgi in situations correlated with high cholesterol amounts [122]. The observation that TM6SF2 possessed the capacity of binding along with stabilization of both ERLIN in addition to APOB, thus act in the form of a connective hub amongst ERLIN in addition to APOB. E167K mutations in TM6SF2 equivalent to deficiencies of TM6SF2 or ERLIN result in aberrations in stabilization of APOB that is one of the crucial factors for the generation of this sub kind of genetic NAFLD [123].
In to these studies emphasized the foundational variation amongst metabolic along with genetic NAFLD. Therefore for patients that are carrying particular SNS areas variable independent molecular pathways might be implicated in rapid NAFLD propagation however further associated with protection conferred to heart. In these cases individualized genotype independent medicine needs clinical applications dependent on the SNS areas presentation, robustness of other symptoms along with of existence other co-morbidities.
Pharmacological Approaches for Treatment of NAFLD along with Reduction in CVD Risk
The basic lifestyle modifications advocated with regards to patients of NAFLD have been diet manipulations along with physical exercise .The diet modifications possessing maximum effectiveness is a low carbohydrate, ketogenic low fat as well as Mediterranean diet with provision of positive action over dyslipidemia, hepatic steatosis, in addition to associated co-morbidities [69, 124]. Akin to that various physical actions (high intensity interval aerobic along with resistance training) have been illustrated to result in reduction of liver fat amounts as well as body weight in addition to recovery of plasma lipid status along with IR. Additionally, these exercises resulted in enhancement in CVD risk factors like plasma amounts of TG-rich VLDL1particles along with LDL-C in addition to reduction of arterial stiffness [125]. An alternative kind of treatment that is efficacious for NAFLD along with NASH in particular when correlated with robust obesity is bariatric surgery whose objective is to result in reduction of food consumption by mechanical means [126]. Furthermore, bariatric surgery results in reduction of risk of CVD processes amidst patients of type2 Diabetes mellitus, as well as obesity hence an akin action would be anticipated in the context of NASH patients [127].
Presently no pharmacological treatments with regards to NAFLD/ NASH has received approval .The treatments that are present have the objective of resulting in reduction of accrual of liver lipids, cause stimulation of metabolic pathways in addition to reduction of liver damage. The major classes of these medicines are:
- Bile acid pathway with metabolism modulators.
- Peroxisome Proliferator Activated Receptor (PPAR)- agonist like Elafibranor.
- Lanfibranor well described drugs for T2mlike Glucagon like peptide 1(GLP-1) Receptor agonist-Semaglutide [128],
- Thyroid hormone Receptor β-agonist (THR-β- agonist) like Resmetriom (MGL-3196B) VK2809 [25, 26] see Figure 5.
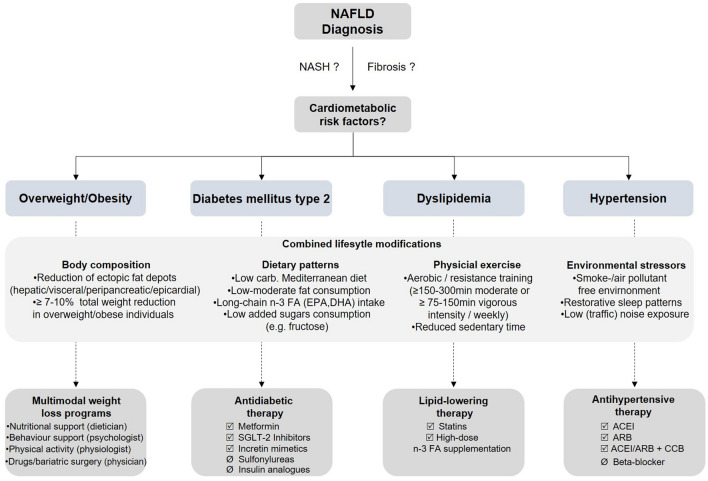
Figure 5: Courtesy ref no-143-A proposed algorithm for management of cardiometabolic risk factors in NAFLD patients. NAFLD non-alcoholic fatty liver disease carb. carbohydrates, n-3 FA omega-3 fatty acids, EPA eicosapentaenoic acid, DHA docosahexaenoic acid, min minute, SGLT2 sodium-glucose linked transporter2, ACEI angiotensin-converting-enzyme inhibitor, ARB angiotensin II receptor blocker, CCB calcium-channel blocker.
Furthermore our concentration is on Fibroblast growth factor 21(FGF21) analogues one of most attractive agent for NAFLD/ NASH therapy possessing the capacity of reduction of risk of CVD which have been corroborated.
FGF21represents an endocrine hormone that belongs to the FGF family, that gets liberated mostly by liver along with demonstrated various metabolic actions. The FGF signaling pathway gets initiated with binding the co- receptors β Klotho (KLB) along with FGF receptor1(FGFR1) that generates FGF21/ FGFR1/KLB complex. This triple complex possesses the capacity of phosphorylation of extracellular signal -regulated kinase1 &2 (ERK1/2) along with Fibroblast growth factor receptor substrate2α (FGFR Sα) with numerous downstream targets [128]. The precise signaling cascade with regards to FGF has not been fully elucidated; however we have insight that in liver it gets controlled by PPARα besides the probability of suppression by liver X receptor (LXR) [129]. FGF21 possesses significant role in the glucose along with lipid metabolism in addition to insulin sensitivity. FGF21 generation occurs primarily in liver in reaction to metabolic cresses, like keto genic diet/fasting besides is the requirement for control of lipolysis, ketogenesis as well as FAO) [130]. Escalation of amounts of FGF21( in Circulation as well as mRNA) have been seen in subjects of robust metabolic conditions (NAFLD, obesity, type2 Diabetes mellitus)that pointed that, it conferred protection against these diseases [131]. Additionally, liberation of FGF21 was possible from the Brown Adipose tissue (BAT) along with take part in thermal control in an ATF4-based manner [132]. Clinical trial of numerous FGF21analogues has corroborated the effectiveness as an attractive therapy for NAFLD.
PF-05231023, that is a long acting analogue of FGF21 have been evaluated in patients with obesity with as well as without T2DM. Significant reduction of TG amounts along with enhancement of HDL-C besides adiponectin was the result of administration of PF-05231023 [133]. Pegbelfermin (BMS-986036), that represented a PEGylated analogue of FGF21 has been assessed in a phase 2a trial in patients of NAFLD along with obesity. 16 weeks of subcutaneous injection of Pegbelfermin caused an important decrease in liver fat [134]. AKR-001, (alias an FGF21analogue) possessed a positive impact on the profile of lipoprotein (TG, n HDL-C, HDL-C, APOB, APOC) in addition to enhancement of insulin sensitivity [135]. In view of the rapid inactivation of FGF21 by hydrolysis, for avoidance of FGF21 cleavage has been tried by hampering its major protease FAP (separate). The assessment of a FAP inhibitor, BR103354 was conducted in vitro in mice along with non-human primate models where it was demonstrated to result in reduction of non-fasting glucose along with TG amounts as well as recovery of hepatic steatosis along with fibrosis. This pointed that FAP inhibitors can act as probable antidiabetic in addition to anti-NASH medicines [136].
The assessment of anti-atherosclerotic action of FGF21 was conducted in various clinical trials where it was illustrated to result in enhancement of cardio metabolic profile in the context of patients of obesity along with T2DM [137]. Significant escalation of lipid profiles as well as reduction of vascular inflammation, amelioration of apoptosis, oxidative stress (OS) besides atherosclerosis associated diseases [138].
Inspite of numerous positive results documented with regards to safety along with tolerance of long term utilization of FGF21, different pernicious actions are existent. In mice FGF21 resulted in hampering of osteoblast genesis through PPARγ hence association of bone turnover as well as energy metabolism [139]. Akin actions on the bone turnover markers were further seen in case of patients with T2DM in humans in the context of a trial for FGF21analogue PF- 05231023 where alterations of N terminal propeptides along with C propeptides cross linking of collagen were observed [133]. Moreover based on dosage till 92% of subjects were in possession of escalation of titers of anti FGF21 antibodies which was a botheration with regards to the immunogenicity if long term NAFLD treatment was attempted with FGF21 as well as its analogues [140]. Nevertheless, it is essential to be aware that inspite of positive action of FGF21 along with its analogues with regards to co-morbidities of NAFLD along with reduction of liver fat a recently conducted systematic study pointed that greater advantageous outcomes were feasible with weight reduction through diet modifications, as well as exercise [141, 142, 143].
Conclusions
The intricate correlation amongst NAFLD along with CVD gets corroborated by the findings that CVD is the commonest etiology of demise amongst NAFLD patients. Liver mitochondrial fatty acids β-oxidation represents the primary system which is responsible for responding to the imbalance with regards to the nutrient inflow. The alterations in the liver lipid metabolism that occur subsequently are the ones implicated in driving towards the generation of NAFLD with concomitant development of a CVD facilitating proatherogenic milieu through a system- wide generation of CK, dyslipidemia, IR along with imbalance in the procoagulant factors. Generation of NAFLD in addition to changes in the complicated networks of genes which possess the capacity of reacting to intracellular as well as environmental stresses, circadian rhythm, nutrients besides lifestyle. Presently numerous treatments with regards to NAFLD/ NASH medicines that are possessing advantageous actions that confer protection to CVD are getting generated. Nevertheless, with the knowledge of distinctive types of metabolic pathways implicated in the pathogenesis of genetic NAFLD a precise insight with regards to the molecular modes are needed for provision of treatments that are efficacious. Inspite of achieving considerable success in getting insight in the context of which NAFLD- CVD result in, a broad range of pharmacological gadgets are accessible, yet lot of work needs to be invested with regards to facilitation, of a healthy life style, nutritional status, status of education along with avoidance of smoking, hence avoiding maximum factors implicated in generation of CVD in NAFLD.
References
-
Kasper P, Martin A, Lang S, Kutting F, Goeser T, et al. (2020) NAFLD and cardiovascular disease: a Clinical review. Clin Res Cardiol 110(7): 921-937.
-
Baratta F, Pastori D, Angelico F, Balla A, Paganini AM, et al. (2020) Nonalcoholic fatty liver disease and fibrosis associated with increased risk of cardiovascular events in a prospective study. Clin Gastroenterol Hepatol 18(10): 2324-2331.
-
Przybysszewski EM, Targher G, Roden M, Cory KE (2021) Nonalcoholic fatty liver disease and cardiovascular disease. Clin Liver Dis 17(1): 19-22.
-
Goncalves DS, Hovingh GK, Nieuwdrop M, Hollebpoom AG (2019) NAFLD and Atherosclerosis: two sides of the same dysmetabolic coin?. Trends Endocrinol Metab 30(12): 891-902.
-
Chiriac S, Stanciu C, Gileanu I, Cojocariu C, Sfarti C, et al. (2021) Non-alcoholic fatty liver disease and cardiovascular disease: the heart of the matter. Can J Gastroenterol Hepatol 6696857.
-
Libby P (2021) The changing landscape of Atherosclerosis. Nature 592(7855): 524-533.
-
Libby P, Buring JE, Badimon L, Hannson GK, Deanfield J, et al. (2019) Atherosclerosis. Nature Rev Dis Primer 5(1): 56.
-
Oni E, Budoff MJ, Zeb I, Li D, Veledar E, et al. (2019) Non- alcoholic fatty liver disease is associated with arterial distensiblity and carotid intima-media thickness(from the multi ethnic study of Atherosclerosis). Am J Cardiol 124(4): 534-538.
-
Chandrasekharan K, Alazawi W (2020) Genetics of Non- alcoholic fatty liver disease and cardiovascular disease: implications for therapy?. Front Pharmacol 10: 1413.
-
Sazanova MA, Ryzhkova AI, Sinyov VV, Galitsyna EV, Melnichenko AA, et al. (2018) Mitochondrial genome mutations associated with myocardial infarction. Dis Markers 2018: 9749457.
-
Dabravolski SA, Bezsonov EE, Baig MS, Popkova TV, Nedusogova LV, et al. (2021) Mitochondrial mutations and Genetic factors determining NAFLD risk. Int J Mol Sci 22(9): 4459.
-
Sliz E, Sebert S, Wurtz P, Kangas A, Soininen P, et al. (2018) NAFLD risk alleles in PNPLA2, TM5SF2, GCKR, and LYPLAL1 show divergent metabolic effects. Hum Mol Genet 27(12): 2214-2223.
-
Simons N, Isaacs A, Koek GH, Kucs, Schaper NC, et al. (2019) PNPLA3, TM6F2 and MBOAT17 genotypes and coronary artery disease. Gastroenterology 152(4): 912- 913.
-
Grimauto S, Pipitone RM, Pennisi G, Celsa C, Camma C, et al. (2020) Association between PNPLA3rs738409C>Gvariant and liver related outcomes in patients with Non-alcoholic fatty liver disease. Clin Gastroenterol Hepatol 18(4): 935-944.
-
Kulvinder KK, Allahbadia GN, Singh MA (2019) Mini Review on Development of Newer Therapies for Non- Alcoholic Fatty Acid Liver Disease with Emphasis on Vitamin D and its Receptor and Allyl Isothiocyanate (AITC). Acta Scientific Nutritional Health 3(12): 1-5.
-
Kulvinder KK, Allahbadia GN, Singh M (2020) An Update on Further Progression of NAFLD, NASH with Prospective Therapies Like L-Carnitine (LC), Nicotinamide Ribose (NR) Combination, as well as Apical Sodium Dependent Bile Acids Transporter (ASBT) or Volixibat and Silybin as Alternatives. Int J Clin Med Cases. 3(3): 138.
-
Kulvinder KK, Allahbadia GN, Singh M (2019) Have Probiotics and Synbiotics passed the test of time to be implemented in management of obesity and related metabolic disorders-a comprehensive review. Adv Obes Weight Manag Control 9(1): 21-28.
-
Kulvinder KK, Allahbadia GN, Singh M (2020) Will Probiotics Provide the Answer for Therapy of Non- alcoholic Fatty Liver Disease (NAFLD)?-A Systematic Review. Biochem Physiol 9(1): 257.
-
Kulvinder KK, Allahbadia GN, Singh M (2019) Rosmarinic Acid-A New Hope for Liver Diseases Like Cirrhosis, Hepatocellular Carcinoma-Needs Translation to Humans. EC Endocrinology and Metabolic Research 4(6): 289-301.
-
Kulvinder KK, Allahbadia GN, Singh M (2020) How do we apply advances in knowledge of Hepatic Macrophages in treating Liver Diseases especially non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepapititis (NASH), with the increasing incidence of Diabesity-A Systematic Review. EC Endocrinology and Metabolic Research.
-
Kulvinder KK, Allahbadia GN, Singh M (2021) Mechanisms that associate extension of Nonalcoholic fatty liver diseases (NAFLD) to NASH (Non-alcoholic steatohepatitis) and further progressing to cirrhosis and Hepatocellular carcinoma(HCC) in addition to few proposed biomarkers for poor prognosis. J Gynaecol 1(16): 1-18.
-
Kulvinder KK, Allahbadia GN, Singh M (2021) How can we optimize therapy of Non-Alcoholic Fatty Acid Liver Disease-A Short Communication on role of Astragaloside IV and other prospective agents. Clinical Research and Clinical Case Reports 1(3): 1-4.
-
Kulvinder KK, Allahbadia GN, Singh M (2015) Therapeutic Applications of the Recent Understanding of Brown or “Beige” Adipocyte Physiology. Adv Tech Biol Med 3: 128.
-
Kulvinder KK, Allahbadia GN, Singh M (2021) Role of Adipocyte impairment in Heart Failure Induction in subjects that are obese along with pre diabetes and overt Diabetes mellitus -A Systematic Review. J Cardiol & Card Disord 2(1): 1-21.
-
Kulvinder KK, Allahbadia GN, Singh M (2021) An update on management of Nonalcoholic Fatty Liver Disease &Nonalcoholic Steatohepapititis-Is the time ripe for achieving resolution of NAFLD &NASH soon. J Endocrinol Res 3(2): 44-60.
-
Kulvinder KK, Allahbadia GN, Singh M (2021) The Association of Non-Viral Liver Diseases from NAFLD to NASH to HCC with the Pandemic of Obesity, Type 2 Diabetes, or Diabesity & Metabolic Syndrome- Etiopathogenetic Correlation along with Utilization for Diagnostic & Therapeutic Purposes-A Systematic review. J Endocrinol 3(2): 10-34.
-
Armandi A, Rosso C, Caviglia GP, Bglanesi E (2021) Insulin resistance across the spectrum of Nonalcoholic fatty liver disease. Metabolites 11(3): 155.
-
Fujii H, Kawada N, Japan Study Group of NAFLD (JSG- NAFLD) (2020) The role of Insulin resistance and Diabetes in Nonalcoholic fatty liver disease. Int J Mol Sci 21(11): 3863.
-
Ryan JD, Armitage AE, Cubbold JF, Banerjee R, Borani O, et al. (2018) hepatic iron is the major determinant of serum ferritin in NAFLD patients. Liver Int 38(1): 164- 173.
-
Haas JT, Vonghia L, Mogilenko DA, Verrijken A, Coste OM, et al. (2019) Transcriptional networks analysis implicates altered hepatic immune function in NASH development and resolution. Nat 1(6): 604-614.
-
Hill MA, Yang Y, Zhang L, Sun Z, Jia G, et al. (2021) Insulin resistance, cardiovascular stiffening and cardiovascular disease. Metabolism 119: 154766.
-
Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri VO, et al. (2020) The Diabetes mellitus- Atherosclerosis connection, :the role of glucose and lipid metabolism and chronic inflammation. Int J Mol Sci 21(5): 1835.
-
Pan X, Kaminga AC, Chen J, Lou M, Lou J (2020) Fetuin-A and Fetuin-B in Nonalcoholic fatty liver disease:a meta- analysis and meta-regression. Int J Environ Res Public Health 17(8): 2735.
-
Liu S, Xiao J, Zhao Z, Wang M, Wang Y, et al. (2020) Systematic review and meta-analysis of Circulating Fetuin A levels in Nonalcoholic fatty liver disease. J ClinTrans Hepatol 9(1): 3-14.
-
Jimenez MC, Sun Q, Schursks M, Hu FB, Manson JE, et al. (2014) Circulating Fetuin A and risk of ischemic stroke in women. Clin Chem 60(1): 165-173.
-
Lichtenauer M ,Wernley B, Paar V, Rohm I, Jung C, et al. (2018) Specifics of Fetuin A levels in distinct types of chronic heart failure. J Clin Lab Anal 32(1): e22179.
-
Lee JH, Lee HS, Cho AR, Lee YJ, Kwon YJ (2021) Nonalcoholic fatty liver disease is an independent risk factor LDL cholesterol target level. IntJEnviron Res Public Health 18(7): 3442.
-
Tutunchi H, Naein F, Maneghami ME, Mobasseri M, Naghashi S, et al. (2021) The association of the steatosis severity, NAFLD fibrosis score and FIB index with atherogenicdys lipidemia in patients with NAFLD: a cross sectional study. Int J Clin Practn 75(6): e14131.
-
Di Costanzo A, Ronca A, DErasmo L, Manfredini M, Bratta F, et al. (2020) HDL mediated cholesterol efflux and plasma loading capacities are altered in subjects with Metabolically-but not genetically driven Non-alcoholic fatty liver disease (NAFLD). Biomedicine 8(12): 625.
-
Adomi MP, Ronda N, Bernini F, Zimetti F (2021) High density lipoprotein cholesterol efflux capacity and Atherosclerosis in cardiovascular disease: pathophysiological aspects and Pharmacological perspectives. Cells 10(30): 574.
-
Gordon SM, Amar MJ, Jeiran K, Stagliano M, Staller E, et al. (2020) Effect of niacin monotherapy on high density lipoprotein composition and function. Lipids Health Dis 19: 190.
-
Aissa AF, Tryndyak V, de Conti A, Melnyk S, Gomes TDUH, et al. (2014) Effect of methionine deficient and methionine supplemented diets on the hepatic one carbon and Lipid metabolism in mice. Mol Nuts Food Res 58(7): 1502-1512.
-
Xu Y, Guan Y, Yang X, Xia Z, Wu J (2020) Association of serum homocysteine levels with, histological severity of NAFLD. J Gastroenterol Liver Dis 29(1): 51-58.
-
Yuan S, Mason M, Carter P, Burgess S, Larsson SC (2021) Homocysteine, B Vitamins, and cardiovascular disease: a Mendelian randomization study. BMC Med 19: 97.
-
Djuric D, Jakovjevic V, Zivkoric V, Srejovic I, et al. (2018) Homocysteine and homocysteine related compounds: anoverview of the roles in the pathology of cardiovascular and nervous system. Can J Pharmacol Physiol 96(10): 991-1003.
-
Fricker ZP, Pedley A, Messaro JM, Vasan RS, Hoffmann U, et al. (2019) Liver fat is associated with markers of inflammation and Oxidative stress in analysis of data from the Framingham Heart Study. Clin Gastroenterol Hepatol 17(6): 1157-1164.
-
Katsarou A, Moustakas II, Pyrina II, Lembessis P, Koutsiliers M, et al. (2020) Metabolic inflammation as an instigator of fibrosis during Nonalcoholic fatty liver disease. World J Gastroenterol 26(17): 1993-2011.
-
Lee Y, Kim K, eun Yoo M, Kim G, Yoon H, et al. (2018) Association of Non-alcoholic Steatohepatitis with myocardialdys function in non- cirrhotic patients. J Hepatol 68(4): 764-772.
-
Chiu LS, Pedley A, Messaro JM, Benjamin EJ, Mitchell GF, et al. (2020) The association of Nonalcoholic fatty liver disease and cardiac structure and function- Framingham Heart Study. Liver Int 40(10): 2445-2454.
-
Hodson L (2019) Hepatic fatty acid synthesis and partitioning: the effect of hepatic partitioning nutritional state. Proc Nutr Soc 78(1): 126-134.
-
Smith JI, Shankaran M, Yoshino M, Schweistzer GG, Chandronikola M, et al. (2020) Insulin resistance drives hepatic de novo lipogenesis in Nonalcoholic fatty liver disease. J Clin Investig 130(3): 1453-1460.
-
Chen Z, Tian R, She Z, Cal J, Li H (2020) Role of Oxidative stress in the pathogenesis of Nonalcoholic fatty liver disease. Free Radical Biol Med 152: 116-141.
-
Chornyl S, Ijlst L, van Roermund CWT, Wanders RJA, Waterham HR (2021) Peroxisomal metabolite and cofactor transport in human. Front Cell Dev Biol 8: 613892.
-
Miotto PM, Petrick HL, Hollowway GP (2020) Acute insulin deprivation results in altered mitochondrial substrate sensitivity conductive to greater fatty acid transport. Am J Physiol Endocrinol Metab 319(2): E345-E353.
-
Dabravolski SA, Bezsonov EE, Baig MS, Popkova TV, Orekhov AN (2021) Mitochondriallipid homeostasis at the crossroads of liver and heart diseases. Int J Mol Sci 22(13): 6949.
-
Brown ZJm, Fu Q, Ma C, Kruhlak M, Zhang H, et al. (2018) Carnitinepalmoyl transferase gene up regulation by linolenic acid induces CD4+T cell apoptosis promoting HCC development. Cell Death Dis 9(6): 620.
-
Fernandes GW, Bocca BMLC (2020) Hepatic mediators of lipid metabolism acid ketogenesis. Focus on fatty liver and Diabetes. Curr Diabetes Rev 17(7): e110320187539.
-
Andany MMA, Freire NC, Filguera MS, Fernandes CF, Bayolo DM (2019) Mitochondrial β- oxidation of saturated fatty acids in humans. Mitochondrion 46: 73- 90.
-
Colbert CL, Kim CW, Moon YA, Henry LA, Patnikar M, et al. (2010) Crystal structure Spot 14, a modulator of fatty acid synthesis. Proc NatlAcad Sci USA 107(44): 18820- 18825.
-
Yang JH, Kim NH, Yun JS, Cho ES, Cha YH, et al. (2020) Snail augments fatty acids oxidation by suppression, of mitochondrial ACC2 during Cancer progression. Life Sci Alliance 3(7): e202000683.
-
Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, et al. (2018) AcetylCoA carboxylase inhibition reverses NAFLD and hepatic Insulin resistance but promotes hyper triglyceredemia in rodents: Hepatology. Hepatology 68(6): 2197-2211.
-
Boudaba N, Marion A, Hurt C, Viollet B, Foretz M (2018) AMPK Re activation, suppresses ion hepapitic steatosis but it’s down regulation does not promote fatty liver development. E Bio Medicine 28: 194-209.
-
Ming Y, Yin Y, Sun Z (2020) Interaction of nuclear receptor subfamily group A, member1 (Nr4a1) and liver kinaseB1 (LKB1) [mitigatetype2 Diabetes mellitus by activating monophosphate activated protein kinase (AMPK)/ Sirtuin 1 (SIRT1 )axis and inhibiting nuclear factor κB (NFκB) activation. Med Sci Monit 26: e92278.
-
Jimenez VM, Espinosa NC, Varela ER, Cardenas MV, Espinoza MB, et al. (2019) Altered levels of Sirtuin genes (SIRT1, SIRT2, SIRT3 and SIRT6) and their target genes in Adipose tissue from individuals with obesity. Diabetes Metab Syndr Clin Res Rev 13(1): 582-589.
-
Kathirvel E, Morgan K, French SW, Morgan TR (2013) Acetyl-l carnitine and lipoic acid improves mitochondrial abnormalities and serum levels of liver enzymes in a mouse model of Non-alcoholic fatty liver disease. Nutr Res 33(11): 932-941.
-
Frigini EN, Barrera EE, Pantano S, Porasso RD (2020) Role of membrane curvature on the activation/de activation of carnitine palmoyl transferase1A: a coarse grain molecular dynamic study. Biochim BiophysActa BBABiomembr Res Commun 1862(2): 183094-183024.
-
Naguib G, Morris N, Yang S, Fryzek N, Williams VH, et al. (2020) Dietary fatty acids oxidation is decreased in Nonalcoholic fatty liver disease: a palmitate breath study. Liver Int 40(3): 590-597.
-
Selen ES, Choi J, Wolfgang MJ (2021) Discordant hepatic fatty acid oxidation and triglyceride hydrolysis leadsto liver disease. JCI Insight 6(2): e135626.
-
Vasan PKL, Dufour S, Lyu K, Zhang XM, Hakarainen A, et al. (2020) Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in Nonalcoholic fatty liver disease. Proc Natl Acad Sci USA 117(13): 7347-7354.
-
Fletcher JA, Deja S, Satapati S, Fu X, Burgess SC, et al. (2019) Impaired ketogenesis and increased Acetyl-CoA oxidation promote hypoglycemia in human fatty liver. JCI Insight 5(11): e127737.
-
Yin HJ, Zhu Y, Malik V, Li X, Peng X, et al. (2021) Intake of sugar sweetened and low calorie sweetened beverages and risk of cardiovascular disease. A meta-analysis. Adv Nutr 12(1): 89-101.
-
Chandra A, Rosjo H, Svensson M, Vigen T, Hansen HI, et al. (2020) Plasma linolenic acid levels and cardiovascular risk factors: results from the Norwegian ACE 1950 study. Eur J Clin Nutr 74(12): 1707-1717.
-
Softic S, Gupta MK, Wang GX, Fujisaka S, Oneil BT, et al. (2017) Divergent effect of glucose and fructose on hepatic lipogenesis and insulin signaling. JClin Investig 127(11): 4059-4074.
-
DiStefano JK (2020) Fructose mediated effects on gene expression and epigenetics mechanism associated with NAFLD pathogenesis. Cell Mol Life Sci 77(11): 2079- 2090.
-
Choi YJ, Shin HS, Choi HS, Park JW, Jo I, et al. (2014) Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP1c, activation in hepatocyte. Lab Investig 94(10): 1114-1125.
-
Weeghel MV, Abdurrachim D, Neidelrof R, Argmann CA, Houtkooper RH, et al. (2018) Increased cardiac fatty acid oxidation in a mouse model with decreased malonyl CoA sensitivity to CPT1B. Cardiovasc Res 114(10): 1324- 1334.
-
Zhang X, Zhang YL, Qiu G, Pian L, Guo L, et al. (2020) Hepatic neddylation targets and stabilizes electron transport flavoprotein s to facilitate fatty acids β - oxidation. Proc NatlAcad Sci USA 117(5): 2473-2483.
-
Kulvinder KK, Allahbadia GN, Singh M (2021) Potential role of Epigenetic Modulation in prevention or therapy for Diabetic Kidney Disease-still a dream or a reality -A Systematic Review. J Diab Nephro Diab Mgmt 1(1): 1-26.
-
Wang C, Zhao Y, Gao X, Li L, Yuan Y, et al. (2015) Perilipin 5 improves hepatic lipotoxicityby inhibiting lipolysis. Hepatology 61(3): 870-882.
-
Jin Y, Tan Y, Chen L, Liu Y, Ren Z (2018) Reactive oxygen species induces lipid droplet accumulation in HepG2 cells by increasing Perilipin 2 expression. Int J Mol Sci 19(11): 3445.
-
Trevino MB, Hurt DM, Machida Y, King T, Nadler J, et al. (2015) Liver Perilipin 5 expression worsens Hepatosteatis but not insulin resistance in high fat diet fed mice. Mol Endocrinol 29(10): 1414-1425.
-
Lin J, Zheng S, Attie D, Keller MP, Bernlohr DA, et al. (2018) Perilipin 5 and Liver fatty acids binding protein function to restore quiescence in mouse hepatic stellar cell. J Lipid Res 59(3): 416-428.
-
Keenan SN, Meex RC, Lo JCY, Ryan A, Nie S, et al. (2019) Perilipin 5 deletion in Hepatocytes remodels lipid metabolism and causes hepatic Insulin resistance in mice. Diabetes 68(3): 543-555.
-
Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, et al. (2011) Perilipin 5, a lipid droplet associated protein provides physical and metabolic linkage to mitochondria. J Lipid Res 52(12): 2159-2168.
-
Yu J, Loh K, Song Z, Yang H, Zhang Y, et al. (2018) Update on glycerol phosphate3acyl transferases:the role in the development of insulin resistance. Nutr Diabetes 8(1): 34.
-
Benador IY, Velliova M, Mahadaviani K, Petchersky A, Wikstrom JD, et al. (2018) Mitochondria bound to lipid droplets have unique bioenergetics expenditure, composition and dynamics that support lipid droplets expansion. Cell Metab 27(4): 869-885.
-
Tan Y, Jin Y, Wang Q, Huang J ,Wu Z, et al. (2019) Perilipin 5 protects against cellular Oxidative stress by enhancing mitochondrial function in HepG2 cells. Cells 8(10): 1241.
-
Asimakopoulou A, Engel KM, Gassler N, Bracht T, Sitek B, et al. (2020) Perilipin 5 protects against hepatic injury in Nonalcoholic fatty liver disease via missing the inflammasome activation. Cells 9(6): 1346.
-
Asimakopoulou A, Vucur M, Luuedde T, Schneiders, Kalampoka S, et al. (2019) Perilipin 5 and lipocalin 2 expression in Hepatocellular carcinoma. Cancers 11(3): 385.
-
Zhou P, Li M, Han X, Bi Y, Zhang W, et al. (2019) Perilipin 5 deficiency promotes atherosclerosis progression, through accelerating inflammation, apoptosis, and Oxidative stress. J Cell Biochem 120(11): 19107-19123.
-
Liu Q, Zhou Z, Liu P, Zhang S (2019) Comparative proteomic study of lipid droplets and mitochondria in mice housed in different temperatures. FEBS Lett 593(16): 2118-2138.
-
Badenes M, Amin A, Garcia IG, Felix I, Burbridge E, et al. (2020) Deletion of iRhom2 protects against diet induced obesity by increasing thermogenesis. Mol Metab 31: 67- 84.
-
Sanchez PBM, Kritzanac M, Weiskirchen R, Asimakopoulou A (2021)Understanding the role of Perilipin 5 in Nonalcoholic fatty liver disease and its role in Hepatocellular carcinoma: a review of novel insights. Int J Mol Sci 22(10): 5284.
-
Chen Z, Qin H, Qiu S, Chen G, Chen Y (2019) Correlation of triglyceride to high density lipoprotein with Nonalcoholic fatty liver disease amongst the non-obese Chinese population with normal lipidlevels: a retrospective cohort research. Lipids Health Dis 18(1): 162.
-
Aicha SB, Badimon L, Vilahur G (2020) Advances in HDL: much more than lipid transporters. Int J Mol Sci 21(3): 732.
-
McGlaughon JL, Pasquali M, Wallace K, Ross J, Cosar OS, et al. (2019) Assessing the evidence for genes implicated in fatty acids oxidation disorders using the validity framework. Mol Genet Metab 128(1-2): 122-128.
-
Di Cristofano M, Mosca AF, Digiacomo M, Fusco C, Boscaino F, et al. (2021) Mechanisms underlying the hermetic effect of conjugated linolenic acid:focus on Nrf2, mitochondria and NADPH Oxidase. Free Radical Biol Med 167: 276-281.
-
Li Q, Tomcic K, Zhang S, Puchowicz A, Zhang GA (2012) Dietary regulation of catabolic disposal of 4hydroxynonenal analogues in rat liver. Free Radical Biol Med 52(6): 1043-1053.
-
Wanders RJA, Viaz FM, Waterham HR, Ferninandusse S (2021) Fatty acids oxidation in Peroxisome: enzymology, metabolic crosstalk with other organelles and Peroxisomal disorders. Advances in experimental medicine and biology 1299: 55-70.
-
Dalhan N, Francisco T, Falter C, Rodriques T, Kalel V, et al. (2021) Current Advances in the and biogenesis of Peroxisomes and their roles in health and disease. Histochem Cell Biol 155(4): 513-524.
-
Bharathi SS, Zhang Y, Gong Z, Mujumdar R, Goetzman ES (2020) Role of mitochondrial acetylCoA dehydrogenases in the metabolism of dicarboxylic Fatty acids. Biochem Biophys Res Commun 527(1): 162-166.
-
Grunig D, Duthaler U, Krahenbuhl S (2018) Effect of toxicants on Fatty acids metabolism in HepG2 cells. Front Pharmacol 9: 257.
-
Lee J, Choi J, Alpergin ESS, Zhao L, Harting T, et al. (2017) Loss of hepatic long chain Fatty acids oxidation confers resistance to diet induced obesity and glucose intolerance. Cell Rep 20(3): 655-667.
-
Lee J, Choi J, Aja S, Scafidi S, Wolfgang MJ (2016) Loss of Adipose Fatty acids oxidation does not potentiate obesity at thermoneutrality. Cell Rep 14(6): 1308-1316.
-
Ramos DC, Mehta R, Solinas CAA (2019) Fibroblast growth factor 21 and browning of White adipose tissue. Front Physiol 10: 37.
-
Nguyen KT, Qiu Y, Cui X, Goh YPS, Mwangi J, et al. (2011) Alternatively activated macrophages produce catecolamines to sustain adaptive thermogenesis. Nature 480: 104-108.
-
Menegaut L, Thomas C, Lagrost L, Masson D (2016) Fatty acid metabolism in macrophages: a target in cardiometabolic diseases. Curr Opin Lipidol 28(1): 19- 26.
-
Nomura M, Liu J, Yu ZX, Yamazaki Y, Yan Y, et al. (2019) Macrophage Fatty acids oxidation inhibts atherosclerosis progression. J Mol Cell Cardiol 127: 270- 276.
-
Wek RC (2018) Role of eIF2 kinases in -translational control and adaptat ion to cellular stress. Cold Spring Harb Perspect Biol 10(7): a32870.
-
Masson GR (2019) Towards a model of GCN2 activation. Biochem Soc Trans 47(5): 1481-1488.
-
Liu S, Yuan J, Yue W, Bi Y, Shen X, et al. (2018) GCN2 deficiency Protects high fat diet induced hepatic Steatosis and insulin resistance in mice. Biochim Biophys Acta Mol Basis Dis 1864(10): 3257-3267.
-
Luo X, Shi X, Sun Z, Ziao J, Song H, et al. (2020) GCN2 deficiency enhances Protective effects of exercise on hepatic Steatosis. Bio Med Res Int, pp: 1-10.
-
Feng W, Leu T, Wang Y, Feng R, Yuan J, et al. (2019) GCN2 deficiency ameliorates cardiac dysfunction in diabetic mice by reducing lipotoxicity and Oxidative stress. Free Radical Biol Med 130: 128-139.
-
Kulvinder KK, Allahbadia GN, Singh M (2020) Synergistic anti-oxidant Actions of Mitochondrial Palatin-like PhosphoLipaseA2Domain-Containing Protein-8(PNPLA8/iPLA2γ) with the Fatty Acids(FA)- Translocating-SLC-25 Gene Family Transporters with their Sequelae-A Review. Int J Med B Health Res 4(1): 1-15.
-
Brouwers MCGI, Simons N, Stehouwer CDA, Koek GH, Schaper NC, et al. (2019) Relationship between nonalcoholic fatty liver disease susceptibility genes and coronary artery disease: Hepatology communications. Hepatol Commun 3(4): 587-596.
-
Krawwczyk M, Porticasa P, Lammet F (2013) PNPLA3 associated Steatohepatitis: towards a gene based classification of fatty liver disease. Semin Liver Dis 33(4): 369-379.
-
Smagris E, BasuRay S, Li J, Huang Y, Lai KY, et al. (2015) Pnpla31148Mknock in mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 61(1): 108-118.
-
BasuRay S, Smagris E, Cohen JC, Hobbs HH (2017) PNPLA3 variant associated with fatty liver disease (1148M) accumulates on lipid droplets by evading ubiquitination. Hepatology 66(4): 1111-1124.
-
Luukonen PK, Nick A, HoltaVyori M, Thiele C, Isokuortti E, et al. (2019) Human PNPLA3-1148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insights 4(16): e127902.
-
Saki S, Saki N, Poustchi H, Malekzadeh R (2020) Assessment of genetic aspects of nonalcoholic fatty liver and premature cardiovascular events. Middle East J Dig Dis 12(2): 65-88.
-
Fan Y, Lu H, Guo Y, Zhu T, Barrio MTG, et al. (2016) Hepatic Transmembrane6 super family member2 regulates cholesterol metabolism in mice. Gastroenterology 150(5): 1208-1218.
-
Wright FA, Bonzerato CG, Sliter DA, Wojkiwicz RJH (2018) The erlin2 T651 mutation inhibits erlin 1/2 complex-mediated 1,4,5 triphosphate receptor ubiquitination and of phosphatidyl inositide 3 3triphosphate - receptor binding. J Biol Chem 293(40): 15706-15714.
-
LiB T, Sun M, Li YF, Wang JQ, Zhou ZMF, et al. (2020) ERLIN TM6SF2-APOB complex destabilizes APOB and contributes to non alcoholic fatty liver disease. PLoS Genet 16(8): e10008955.
-
Moradie FS, Cuthbrton DJ, Barrett M, Jackson NC, Herring R, et al. (2016) Exercise trainingreduces Liver fat and increases rates of VLDL clearancebut not VLDL production in NAFLD. J Clin Endocrinol Metab 101(11): 4219-4228.
-
Lassaily G, Caiazzo R, Wandji LCN, Gremmi V, Baud G, et al. (2020) Bariatric surgery provides long term resolution of Nonalcoholic hepatic Steatohepatitis and regression of fibrosis. Gastroenterology 159(4): 1290- 1301.
-
Amiinian A, Zajichek A, Arterburn DE, Wolski KE, Brethauer SA, Schauer PR, et al. (2018) Association of metabolic surgery with major adverse cardiovascular outcomes in patients with type2 Diabetes and obesity. JAMA 322(13): 1271-1282.
-
Deprince A, Haas JT, Steels B (2020) Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab 42: 101092.
-
Jensen Cody SO, Potthoff MJ (2021) Hepatokines and metabolism: deciphering communication from the liver. Mol Metab 44: 101138.
-
Smati S, Regnier M, Fougeray T, Polizzi A, Fougerat A, et al. (2020) Regulation of Hepatokine Gene expression in response to fasting and feeding: influence of PPAR α and insulin dependent signaling In hepatocyte. Diabetes Metab 46(2): 129-136.
-
Fisher FM, Flier EM (2016) Understanding the physiology of FGF21. Annu Rev Physiol 78: 223-241.
-
Barb D, Briil F, Kalavalapalli S, Cusi K (2019) Plasma Fibroblast growth factor 21 is associated with Nonalcoholic hepatic Steatohepatitis with obesity and type2 Diabetes mellitus. J Clin Endocrinol Metab 104(8): 3327-3336.
-
Pereira RO, Marti A, Olvera AC, Tadinada SM, Bjorkman H, et al. (2021) OPA1deletion in brown Adipose tissue improves thermoregulation and systemic metabolism via FGF21. eLife 10: e66519.
-
Kim AM, Somayaji VR, Dong JQ, Rolph TP, Weng Y, et al. (2017) Once weekly administration of a long acting Fibroblast growth factor 21 Analogue modulateslipids, Bone turnover, blood pressure and body weight, differently in obese people with hypetriglyceridemia and in nonhuman primates. Diabetic Obese Med 19(12): 1762-1772.
-
Sanyal A, Charles ED, Tetri BAN, Loomba R, Harrison SA, et al. (2018) Pegbelfermin (BMS-986036), a PEGylated Fibroblast growth factor 21 Analogue in patients with Nonalcoholic hepatic Steatohepatitis: a randomized double blind, placebo controlled phase 2a trial. Lancet 392(10165): 2705-2717.
-
Kaufman A, Abuqayyas L, Denney WS, Tillman EJ, RolphT (2020) AKR-001, An Fc- FGF21Analogue showed sustained Pharmacodynamic effects on insulin and lipid metabolism in type2 Diabetes mellitus patients. Cell Rep Med 1(4): 100057.
-
Cho JM, Yang FH, Quan W, Nam FH, Cheon H (2020) Discovery of a novel Fibroblast activation protein (FAP) inhibitor,BR103354 with antidiabetic and anti steatotic effects. Sci Rep 10: 21280.
-
Krokinnos J, Tang S, Rye KA, Ong KL (2017) The role of Fibroblast growth factor in 21 Atherosclerosis. Atherosclerosis 257: 259-265.
-
Tabbari ES, Karimian A, Parsian H, Rameshknia V, Mahmoodpour A, et al. (2019) The roles of FGF21in Atherosclerosis . Rev Endocrinol Metab Disord 20(1): 103-114.
-
Wei W, Dutchak PA, Wang X, Ding X, Wang X, et al. (2012) Fibroblast growth factor 21 promotes Bone loss by potentiating the effects of Peroxisome Proliferator adenine Activated Receptor γ. Proc NatlAcad Sci USA 109(8): 3143-3148.
-
Charles ED, Tetri BAN, PabloFrias J, Kundu S, LuoY, et al. (2019) Pegbelfermin (BMS-986036), FGF21in patients with Obesity and type2 Diabetes: results from a Randomized Phase 2 Study. Obesity 27(1): 41-49.
-
Kenneally S, Sier J, Moore JB (2017) Efficacy of dietary and physical activity intervention in nonalcoholic fatty liver disease: a Systematic review. BMJ Open Gastroenterol 4(1): e000139.
-
Batchuluun B, Pinkosky SL, Steinberg GR (2022) Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat RevDrug Discov 21(4): 283-305.
-
Kasper K, Martin A, Lang S, Kutting F, Goeser S, et al. (2021) NAFLD and cardiovascular disease: a clinical review. Clin Res Cardiol 110(7): 921-937.
- Shaping Healthy Futures: Pediatric Endocrine Breakthroughs of 2025
- Precision Medicine in Obesity: Customizing Treatment for 2025
- The Thyroid Revolution: How 2025 is Redefining Hormone Health
- Editorial- Targeting Immunometabolism for Generating Innovative Therapies for Cancer
- Current Knowledge of Chickenpox
- Correlation of Preinjection Values of Gonadotropins and Estradiol Level with Clinical and Radiologic Evidence of Sufficient Pubertal Suppression in Girls with Central Precocious Puberty