Xia-Gibbs Syndrome- A Case Report
Background: Xia-Gibbs syndrome a phenotypically heterogeneous genetic neurodevelopmental disorder (NDD) characterized by intellectual impairment with absent poor expressive language, global developmental delay, hypotonia, feeding problems, distinctive mild dysmorphic facial features, is caused by newly arising mutations (including some missense variants) in the AHDC1 gene. Case Presentation: A 10-month-old female child presented with complaints of fever in the past 7 days, respiratory distress for 4 days, failure to thrive, hypotonia, delayed motor milestones, dysmorphic features and global developmental delay. Whole Genome Sequencing revealed AHDCI chr1:27878230G>A-the missense variant NM_001029882.3 (AHDC1):c397C>T (p.Arg133Cys) and phenotype consistent with Xia Gibbs Syndrome. Conclusions: possible pathogenicity of newly observed missense variants/missense changes outside the clusters in AHDC1 in individuals with XGS-like phenotypes can be established with an improved understanding of AHDC1 structure and function.
Abbreviations
XGS: Xia- Gibbs Syndrome; MIM: Medical Image Merge; NDD: Neuro Development Disorder; AHDC1: Associated With The Xia- Gibbs Syndrome; MAF: Minor Allele Frequency; SMA: Spinal Muscular Atrophy; SMN: Survival Motor Neuron; 2 DECHO: 2D Echocardiography; FT: Full Time; CT: Computed Tomographic Angiography; MRI: Magnetic Resonance Imaging; MRS: Magnetic Resonance Spectroscopy; CPK: Creatine Phosphokinase; TNFRSF13B: Tumor Necrosis Factor Receptor; CVID: Common Variable Immunodeficiency; UCMD1: Ulrich Congenital Muscular Dystrophy-1; DNA: Deoxyribonucleic Acid; VOUS: Visual Analogue Scale; OMIM: Online Mendelian Inheritance in Man; ACMG: American College of Medical Genetics And Genomics; VI: Visual Impairment.
Introduction
Xia-Gibbs syndrome (XGS; MIM: 615829), a phenotypically heterogeneous genetic neurodevelopmental disorder (NDD) is caused by truncated (abnormally short) AHDC1 protein synthesis resultant of mutations in the AT-Hook DNA-Binding Motif-Containing 1 (AHDC1) gene on chromosome 1p36 [1]. Fan Xia and Richard A. Gibbs described XGS for the first time in 2014 [2]. More than 270 individuals have been diagnosed with XGS worldwide [3]. Trio exome sequencing identified new pathogenic AHDC1 mutations in most of the cases, many likely variants in other genes were not identified/excluded due to inappropriate genotype-phenotype correlation [2, 4, 5].
Many cases of XGS also have obstructive sleep apnea and brain abnormalities in addition to characteristic features [6]. AHDC1 is intolerant to missense variation as indicated by a positive missense Z score of 2.86 and a missense observed- versus-expected mutation ratio of 0.75. It’s well conserved across most vertebrates, with 94% identity between human and mouse proteins. There are many known rare and ultra- rare AHDC1 variants reported in the Genome Aggregation Database (gnomAD v.2.1.1) including 528 missense variants, of which 98% (518) have a minor allele frequency (MAF) <
0.001 [7].
We report a female child, 10 months of age with AHDCI chr1:27878230G>A, the missense variant NM_001029882.3 (AHDC1):c397C>T (p.Arg133Cys) and phenotype consistent with Xia Gibbs Syndrome.
Case Presentation
A 10-month-old female child was admitted with fever in the past 7 days, respiratory distress for 4 days, failure to thrive, hypotonia, delayed motor milestones, dysmorphic features and global developmental delay. At birth, the first-born full- term (birth weight - 2.5 kg) female child delivered vaginally out of a non-consanguineous marriage was discharged at live day 3. She presented at 4 months of age with, acute diarrhea with some dehydration, bronchopneumonia with hypotonia; clinically suspected to have congenital myopathy.
The patient had a history of recurrent respiratory distress and pneumonia and was evaluated for hypotonia. SMA was ruled out by gene studies. SMN Gene analysis was normal. No deletion/ duplication was noted in exon 7 or exon 8 of the SMN 1 gene; though heterozygous duplication of exon 7 and exon 8 in the SMN2 gene was noted in the clinical sample tested.
On examination, the child had pallor but no icterus, cyanosis, clubbing or lymphadenopathy. Anterior fontanelle was at level. Her weight was 6.4 kg (0.8 centile = <3rd centile), length 58cm and head circumference was 41cm (0.7 centile = <3rd centile). She has no history of seizures. Strabismus, abnormal facies with downward slanting palpebral fissures with partial ptosis of the left eye, horizontal eyebrows, flat nasal bridge, thin upper lip, and low set ears were noted on general physical examination (Figure 1).
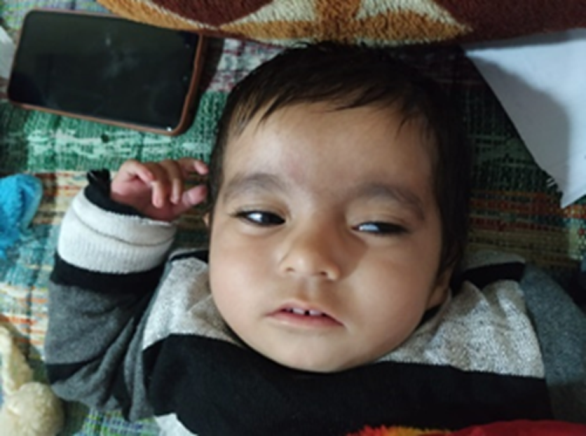
Respiratory examination suggested bilateral crepitations, wheeze and stridor. The child had normal sensorium. Cardiovascular examination and 2 DECHO were normal.
During developmental assessment, significant gross motor, cognitive, verbal and social & emotional delay was noted. On gross motor milestone assessment, pull to sit was absent, partial neck holding was present (coinciding to 2 months age), and sitting with support was absent. Fine motor examination revealed unidextrous grasp and transferring objects positive (coinciding to 8 months of age). On verbal assessment, cooing was present (coinciding to 2 months of age). On social milestone evaluation; recognizes mother and stranger anxiety was present (coinciding to 7 months of age).
Features suggestive of Xia Gibbs syndrome were hypotonia, significant verbal and motor delay, strabismus, abnormal facies, stridor ( laryngomalacia) and genome sequencing.
Complete blood counts revealed total leucocyte counts of 34.59 x103/Cu mm, heamoglobin 7.4 gm/dl with microcytic hypochromic anaemia and platelet counts 810 x 103/Cumm. Differential Leucocytes Counts showed 92% neutrophils with 3%. Band forms. F T 3 was 2 Pg/ml, FT 4 was 7.6 Pg/ ml with TSH-1.22 Uiu/ml. Serum Calcium was 8.9 mg/dl, Vitamin D3 was 141 ng/ml. sodium was 1.31 mEq/L with normal potasium and chloride. CPK Nac was 47u/L. Serum urea and creatinine were normal. In immunoglobulin assay by immunoturbiditimetry with serum; IgA level was 109 mg/dL, IgG - 578 & IgM 72 mg/Dl (normal).
CT Chest suggested patchy areas of consolidation involving subpleural regions of posterior segments of the right upper lobe and posterior basal segment of the right lower lobe. Subpleural interstitial thickening in bilateral lung parenchyma; the posterior segment of the left upper lobe and superior segment of the left lower lobe- findings were suggestive of infective aetiology. MRI brain with contrast was normal; MRS suggested subtle choline-raised rest of the brain metabolite peak are normal - consistent with age. However bilateral mastoiditis, bilateral ethmoids and bilateral adenoids enlarged in the nasopharynx were noted.
Whole genome sequencing suggested AHDCI chr1:27878230G>A with the missense variant NM_001029882.3 (AHDC1):c397C>T (p.Arg133Cys) and this combined with phenotype ascribed to a diagnosis of Xia Gibbs syndrome (Table 1).
| Gene & Transcript | Variant | Location | Zygosity | Disorder (OMIM) | Inheritance | Classification |
|---|---|---|---|---|---|---|
| AHDC1 NM_001029882.3 | c397C>T (p.Arg 133 Cys) | Exon 3 | Heterozygous | Xia-Gibbs syndrome -615829 | Autosomal Dominant | Uncertain Significance |
Table 1: Variant Interpretations in Relation to Findings Related to Phenotype.
AHDCI chr1:27878230G >A Uncertain Significance
The missense variant NM_012452.3 (TNFRSF13B):c241C>A (p.His81Asn) has not been reported previously as a pathogenic variant nor as a benign variant, to our knowledge. The p.His81Asn variant is observed in 2/30,616 (0.0065%) alleles from individuals of gnomAD. South Asian background in gnomAD. The p.His81Asn variant is novel (not in any individuals) in 1Kg. There is a small physicochemical difference between histidine and asparagine, which is not likely to impact secondary protien structure as these residues share similar properties. For these reasons, this variant has been classified as uncertain significance.
Discussion
There is overlap between the clinical manifestations of XGS and those of other rare heterogeneous NDD syndromes, so diagnostic testing by DNA sequencing is required to establish whether a pathogenic or likely pathogenic variant has been identified in AHDC1 [2]. In our case, immunoglobulin levels were normal, ruling out Common Variable Immunodeficiency (CVID), a condition characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections, and an inability to mount an antibody response to antigens caused by a failure of B-Cell differentiation and impaired immunoglobulin secretion. The Ulrich congenital muscular dystrophy-1 (UCMD1) is caused by mutations in three genes encoding collagen type VI subunits (COL6A1, COL6A2 and COL6A3). Symptoms include generalized muscle weakness, striking hypermobility of distal joints, and variable contractures of proximal joints; the condition was ruled out based on nonplausible phenotypes.Individuals with missense mutations and those with truncating mutations show the same overall pattern when phenotypes are compared.
Evaluation of XGS trait manifestations in comparison to well-established pathogenic AHDC1 truncating variant alleles and to other Mendelizing disorders of the AHDC1 locus concludes that some novel missense variants in AHDC1 can cause XGS [1].
Clinical review of the four probands with AHCD1 truncating mutations suggested similar history of congenital hypotonia and history of obstructive sleep apnea, potentially because of upper-airway structural abnormalities [2]. In our case, features suggestive of Xia Gibbs syndrome were hypotonia, significant verbal and motor delay, strabismus, abnormal facies and stridor (laryngomalacia), substantiated by whole genome sequencing.
Five likely pathogenis missense variants in AHDC1 have been reported in the literature with phenotypic similarity with previously reported XGS cases as evidence supportive of pathogenicity. One individual with craniosynostosis was reported with an AHDC1 de novo frameshift mutation (p.C791fs∗57). Five other XGS individuals didn’t had craniosynostosis in the cohort with de novo frameshift mutations at this position [8]. One variant reported by GeneDx indicates a possible XGS diagnosis with a de novo change at position 487 (Arg487Trp) [9]. Correlation with clinical phenotyping and expected observations may be useful in molecular diagnostic interpretation [10].
Conclusion
Ultimately, our study provides insight into the possible pathogenicity of newly discovered missense variants in AHDC1 in individuals with XGS-like phenotypes and in the absence of other genomic variants that might explain one’s clinical presentation. As AHDC1 structure and function are better understood, some missense changes outside the clusters may be identified as pathogenic.
Recommendations
The interpretation of this result should be done in the context of this individual’s clinical and biochemical profile. Variants identified were classified as VOUS based on and as per ACMG guidelines. Prenatal testing is not recommended due to limited or conflicting evidence of association with the disease. If the variant is reclassified in the future as pathogenic or likely pathogenic variants, prenatal testing or pre-implantation genetic diagnosis may be available. Recommendations for prenatal testing must be discussed with a clinical geneticist or a genetic counselor. Genetic counseling is recommended.
References
-
Khayat MM, Hu J, Jiang J, Li H, Chander V, et al. (2021) AHDC1 missense mutations in Xia-Gibbs syndrome. Human Genetics and Genomics Advances 2(4): 100049.
-
Xia F, Bainbridge MN, Tan TY, Michael FW, Angela ES, et al. (2014) De novo truncating mutations in AHDC1 in individuals with syndromic expressive language delay, hypotonia, and sleep apnea. Am J Hum Genet 94(5): 784- 789.
-
He P, Yang Y, Zhen L, Li DZ (2020) Recurrent hypoplasia of corpus callosum as a prenatal phenotype of Xia-Gibbs syndrome caused by maternal germline mosaicism of an AHDC1 variant. Eur J Obstet Gynecol Reprod Biol 244: 208-210.
-
Ordonez LD, Montano DR, Candelo E, Cruz S, Pachajoa H (2019) Syndromic Intellectual Disability Caused by a Novel Truncating Variant in AHDC1: A Case Report Iran. J Med Sci 44: 257-261.
-
Jiang Y, Wangler MF, McGuire AL, Lupski JR, Posey JE, et al. (2018) The phenotypic spectrum of Xia-Gibbs syndrome Am. J Med Genet A 176(6): 1315-1326.
-
Gumus E (2020) Extending the phenotype of Xia- Gibbs syndrome in a two-year-old patient with craniosynostosis with a novel de novo AHDC1 missense mutation. Eur J Med Genet 63(1): 103637.
-
Karczewski KV, Francioli LC, Tiao G, Cummings BB, Alfoldi J, et al. (2020) Genome Aggregation Database Consortium The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581: 434-443.
-
Mubungu G, Makay P, Boujemla B, Yanda S, Posey JE, et al. (2021) Clinical presentation and evolution of Xia-Gibbs syndrome due to pGly375ArgfsTer3 variant in a patient from DR Congo (Central Africa). Am J Med Genet A 185: 990-994.
-
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, et al. (2009) DECIPHER: Database of Chromosomal Imbalance and Phenetype in Humans Using Ensembel Resources. Am J Hum Gen 84(4): 524-533.
-
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, et al. (2017) Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation N Engl J Med 376(1): 21- 31.
- Understanding Pediatric Multiple Sclerosis: Clinical Presentation, Diagnostic Criteria, Therapeutic Advances, and Supportive Care Approaches
- Hemophilia in Children
- A Study to Assess Effectiveness of Play Therapy in Reducing Post-Operative Pain among Children Age 2 To 5 Year who have Undergone General Surgeries in Selected Pediatric Hospitals of Vadodara
- Preterm Birth: Scope of the Problem, Cost of Care, Potential Complications and Current Guidelines for Management
- Noradrenaline: Can we Use it to Manage Hemodynamic Instability among Neonatal Septic Shock at the NICU?
- Occurrence of Third root (supernumerary / Radix Entomolaris / Radix Paramolaris) in the Primary Mandibular Molars - A Systematic Review