Gibbs Free Energy Implications of Electric Double Layer for Wettability Trend in Geologic Carbon Storage
In this paper, we demonstrated the theoretical relationship between Gibbs free energy of the electric double layer and aqueous solution pH. The basis for our theoretical derivation is the fundamental electrochemical equation of Nernst and the Gouy Chapman theory of the electric double layer. In the thermodynamic sense, the partial derivative of Gibbs free energy with regard to interface/surface area is shown to directly linked to interfacial or surface energy, where the former is pH dependent. Accordingly, our theoretical derivation has permitted the interpretation of wettability trends in saline aquifers under geological carbon storage.
Introduction
Electrostatic phenomena are ubiquitous in geologic systems characterized by aqueous-solid interfaces [1] and they are governed by the surface chemistry of solid surfaces and the ambient pH of the aqueous-rock system. The specific surface chemistry of rock surface reflects the mineralogical composition and surface functional groups [2]. Pertinent to the surface chemistry of rocks and or minerals is the point of zero charge pH [3] which is fundamental to the development of the electric double layer system [4]. Next to the surface chemistry and pH that are crucial for electrical phenomena in geologic systems, the interfacial free energy of the solid- liquid interface is fundamental to wettability evolution in multi-phase flow in porous media and this impacts wettability [5] where pH evolves.
Gibbs free energy of surface or interfacial chemical reactions such as protonation or deprotonation of surface ionizable groups is a thermodynamic measure of its feasibility [6] and will govern surface charging reactions under varying pH of aqueous solution. Therefore, under geological sequestration of carbon dioxide in saline aquifers, pH induced surface charge regulation which affects electric double layer development [7] will also affect solid-liquid interfacial tension [8] and wettability. Consequently, pH evolution will also affect the Gibbs free energy of the electric double layer [9]. In the literature, mentions have been made regarding surface charge and potential dependence of the electric double layer and the control of pH on the electric double layer [7], but to the best of our knowledge, a direct relationship between the free energy and pH that is theoretically driven is lacking. Moreover, in the field of colloid science, the electric double layer theory treats the thermodynamics related to the electrostatic Gibbs free energy [10]. This concept can be exploited for the derivation of the electrical or electrostatic Gibbs free energy related to surface charging of aquifer rock surfaces in response to pH changes of formation water. Therefore, the objective of this paper is to theoretically explore the effect of pH changes on Gibbs free energy of electrostatic charging in geologic system and to further use this as the basis for predicting wettability evolution trends in saline aquifers subjected to anthropogenic carbon dioxide injection, given the effect of pH evolution on solid-liquid interfacial tension and its appearance in Young’s phenomenological equation [11].
Background
Dependence of Surface Charge on Aqueous Solution pH
For a siliciclastic saline aquifer with predominant silica composition, the predominant surface acidic functional group is Silanol [12]. This has a surface density [13] determined by the type of silica. These functional groups will ionize by protonation and deprotonation reactions in accordance with the following equations:
$$ \equiv S i O H + H ^ {+} = \equiv S i O H _ {2} ^ {+} \tag {1} $$
$$ \equiv S i O ^ {- 1} + H ^ {+} = \equiv S i O H \tag {2} $$
In which SiOH ≡
is a neutral surface species (Silanol), H + is
hydrogen ion, 2 SiOH + ≡
is protonated surface species,
$$ \equiv S i O ^ {- 1} $$
is deprotonated surface species.
Reaction 1 is typical for silica surface under pH less than the point of Zero charge pH of silica surface while reaction 2 is typical for pH greater than the point of zero charge pH. Generally, these reactions will lead to surface charge development such that the concentrations of charge type will be governed by aqueous solution pH as follows [14]:
2 $$ = \frac {\left[ H ^ {+} \right] _ {p z c} ^ {2}}{\left[ H ^ {+} \right] _ {p z c} ^ {2} + \left[ H ^ {+} \right] _ {p} ^ {2}} $$ + H R H H − pzc pH (3)
2 2 + + pzc pH
2 $$ = \frac {\left[ H ^ {+} \right] _ {p H} ^ {2}}{\left[ H ^ {+} \right] _ {p z c} ^ {2} + \left[ H ^ {+} \right] _ {p} ^ {2}} $$ + H R H H + pH pH (4)
2 2 + + pzc pH In which pH R −is the fraction of surface site available for the adsorption of positive ions from the electric double layer at prevailing bulk solution pH, pH R + is the fraction of surface site available for the adsorption of negative ions from the electric double layer at prevailing bulk solution pH, H + hydrogen ion concentration mol/l, pzc H + is the hydrogen pH H + ion concentration at the point of zero charge pH and is the hydrogen ion concentration at aqueous solution pH
Relevance to Interface Thermodynamics
The surface free energy of a surface is the work done or required to create unit area for a surface in vacuo and the work done or required to create unite interface area where two different phases are in contacts. The origin of this energy has intermolecular bearing [15, 16]. In this regard, the interfacial tension between two phases is given as Oss, et al. [17]:
2 1/2 1/2 ( ) ( )
= − + + − −
γ γ γ LW LW ij i j (5)
( ) ( ) ( ) ( )
1/2 1/2 1/2 1/2 2 γ γ γ γ γ γ γ γ + − + − + − − +
i i j j i i i j In which ij γ is interfacial tension between phase i and phase j [N/m], LW iγ is Lifhitz-van der Waals component of phase i surface tension [N/m], LW jγ is Lifhitz-van der Waals component of phase j surface tension [N/m], iγ + is the electron acceptor component of phase I surface tension [N/m], iγ − is the electron donor component of phase i surface tension [N/m], jγ − is the electron donor component of phase j [N/m] and jγ + is the electron acceptor component of phase j surface tension [N/m]
In geologic systems and considering induced surface charging, the electron acceptor and donor contributions to individual surface tensions originate from reactions 1 and 2 respectively. The thermodynamic implication is that the solid-liquid interfacial tension can be written as:
2 1/2 1/2 ( ) ( )
= − + + − −
γ γ γ LW LW sl s l (6)
( ) ( ) ( ) ( )
1/2 1/2 1/2 1/2 2 γ γ γ γ γ γ γ γ + − + − + − − +
s l l l s l s l In which sl γ is interfacial tension between solid and liquid [N/m], LW iγ is Lifhitz-van der Waals component of solid phase[N/m], LW jγ is Lifhitz-van der Waals component of of liquid phase [N/m], iγ + is the electron acceptor component of solid phase [N/m], iγ −is the electron donor component of solid phase [N/m], jγ −is the electron donor component of liquid phase [N/m] and jγ + is the electron acceptor component of liquid phase [N/m].
Equation 6 and Equation 3 through 4 put together show the interfacial tension between solid and liquid interface in geologic systems will be pH dependent as will the process of surface charging and electrostatic free energy of charging.
Gibbs Free energy of Surface Charging Reactions in Geologic Systems
If pH changes of aqueous solution and the resulting adsorption or desorption [18] of potential determining ions are regarded as surface charging processes, then the Gibbs free energy of such a surface charging reaction will provide an appropriate thermodynamic measure of the feasibility of the process. Since adsorption or desorption of potential determining ions from surface hydroxyl functional groups provide potential sites for the adsorption of counter ions and the charging of surfaces, the electrostatic Gibbs free energy will also provide a means of determining the feasibility or extent of surface free energy changes of aquifer rock surfaces because the surface free energy of solids are changed due to the adsorption of ions or molecules [19]. The electrostatic energy of charging is pH dependent in accordance with the following equation [20]:
( ) ( ) 1, 2.3 el B G pH pH k T Q pH dpH = ∆∫ (7)
Here B k is Boltzmann’s constant, T is the temperature, Q ∆ is the change in average charge and pH is negative logarithm to base 10 of hydrogen ion concentration. 1 pH is a reference pH taken as zero [21].
The physical validity of Equation 7 stems from the fact that a charged surface has none zero electrostatic potential which can give rise to electrostatic interaction energy. In addition, the total electrical Gibbs free energy of the electric double layer is given as:
( ) ( )
0 0 0 0 0 0 f d d σ ψ ψ σ σ σ ψ σ ψ ψ = − = − ∫ ∫ (8)
0
0 , , f ψ σ are electrical Gibbs free energy of the diffuse layer [J/m2], potential of the diffuse layer [V] and surface charge density respectively [C/m2]. Equation 8 was defined for a biological system (proteins) so its analogy will be sought for ionizable groups related to geologic [22] system where surface charge densities can be defined. In this regard, a charge surface has electrostaic potential which gives rise to an electrostatic energy. The relationship between surface charge density and surface potential is given as Atkinson, et al. [18]:
e k T nk T ψ π σ ε = (9)
2 B B
B , ,k , , , , T n e σ ψ ε are surface charge density [C/m2], surface potential [V], Boltzmann constant [J/K], temperature [K], number density of ions [mols/l], dielectric permittivity [F/m] and electronic charge respectively [C]
The solid-liquid interface is usually considered to be made up of three layers [23]. The mineral surface is characterised by the presence of the silanol groups with an electrical potential. The stern layer corresponds to the adsorbed cation and it is characterised by the nature of the interaction between the adsorbed ions and the surface site. The third is the electrical double layer also called Guo layer which is characterised by an internal electrical potential. If the ion density in the diffuse layer obeys the Boltzmann distribution [19] in the electric field created by the ionization of the surface silanols groups, then the diffuse layer charge density can be linked to the diffuse layer potential as Wirth, et al. [21]; Johnson, et al. [24]:
ψ σ ε ε =
ze k T I k T
d B r B
0 8 sinh 2 (10)
0 , , , , , , d B r k T I z σ ψ ε ε Surface charge density [C/m2], potential of the diffuse layer [V], Boltzmann constant. Temperature, relative permittivity [F/m], permittivity of vacuo, ionic strength [mol/l] and charge on ion respectively. All other parameters have their units already defined. Assuming the following [23]:
0 s d ψ ψ ψ = = (11)
0, , s d ψ ψ ψ are solid or mineral surface potential, stern layer potential and diffuse layer potential respectively.
Equation 10 can be written as:
ψ σ ε ε =
ze k T I k T
d B r B
0 8 sinh 2 (12) The validity of this assumption stems from the fact that under certain conditions, the stern layer may contain more counter ions than is required to balance the surface charge in which case the diffuse layer is charged to the same extent as the surface and would therefore have the same potential [25].
The quantity in the bracket expressing the argument of hyperbolic sine in Equation 12 has the following ratio:
ze k T ψ
0 2 B
(13) This is a constant for a given salinity and temperature of aqueous solution. The coefficient of the hyperbolic function is also a constant. This equation can be written as:
( ) 0 0 Sinh A B σ ψ = (14)
Where: 0 8 B r A k T I ε ε = (15)
=
ze B k T
(16)
2 B
The Nernst equation is given as:
( ) 2.303 2.303 B B pzc k T k T pH pH pH e e ψ = − ∆ = − (17)
ψ is surface potential, B k is Boltzmann constant, pH is the negative logarithm to base 10 of the hydrogen ion concentration of the aqueous solution, pzc pH is the point of zero charge pH of the solid surface. Substitution for surface potential from Equation 17 using Nernst equation gives:
( ) ( ) 0 sinh 0.5*2.303* pzc A z pH pH σ = − (18) The pH derivative of surface potential is given as:
0 2.303 B k T d dpH e ψ = − (19)
Substitution into Equation 8b give:
| 2.303k T B | 8k Tε εI B r o |
|---|---|
sinh 0.5 .2.303 pzc To proceed further geological sequestration of anthropogenic carbon dioxide in a siliciclastic saline aquifer with a predominant silica composition and silica cement similar to quartz arenites [26] will be assumed. In this regard, the point of zero charge pH of silica will be 3 averagely [27]. By further assuming a sodium chloride dominated formation brine (HANOR) the ionic charge z is 1. Furthermore, if the surface of silica is implied under typical formation brine pH which is near neutral [28] then the surface of silica with an average point of zero charge pH of 3 will normally develop negative surface charge [28] when subjected to carbon dioxide injection. This means that the surface potential in Equation 8 will be negative [29].
To calculate electrostatic Gibbs free energy, information about aqueous solution salinity, temperature and physical constants given in Equation 23 are required. With regard to salinity Hannor [30], gave a classification of sedimentary basin salinity. One of his classifications recognized under saturated sodium chloride dominated brine with salinities in the range 10000 to 250000 ppm. To calculate Gibbs free energy for a specific case the median value of this range (130000 ppm) will be used. This gives 2.22 moles per litter of sodium chloride. This corresponds to 2.22 normal sodium chloride solution.
The static dielectric constant of brine as a function of temperature and normality is given as Stogryn [31]:
( ) ( ) ( ) ( )
ε ε − − =
1.000 0.2551 5.151*10 6.889*10 a N N N N
( ) 0 , T N ε = is the dielectric permittivity at a given temperature and normality- Fm-1 ( ) 0 ,0 T ε = Dielectric permittivity at a given temperature at zero salinity-Fm-1 is the normality.
The dielectric constant of pure water as a function of temperature is given as Malmber & Maryott [32]: ( ) 4 2 6 3 0 ,0 87.74 0.40008 9.398*10 1.410*10 T T T T ε − − = − + + (22) T = is temperature (oC) To simplify the calculations, this salinity will be taken as that typical of depths of geological carbon sequestration which are normally greater than 800 meters. Assuming a depth of 1000 meters and a mean geothermal gradient of 25oC per kilometer and a mean surface temperature of 10o C, the temperature will be 35.5 oC. Putting the equations together, the dielectric constant of water will be 28 F/m, which substitute for the product of relative permittivity and dielectric constant in Equation 20. Substitution of Boltzmann constant-1.38 *10-23 kJK-1 [33] and integration of Equation 20 gives:
( ) 1.196 cosh 3.45 1.15 f pH = − − + (23) Theoretical Plot
Figure 1 represents a plot with pH range starting from average ambient formation water pH [28] to pH values slightly below the point of zero charge pH of silica surface.
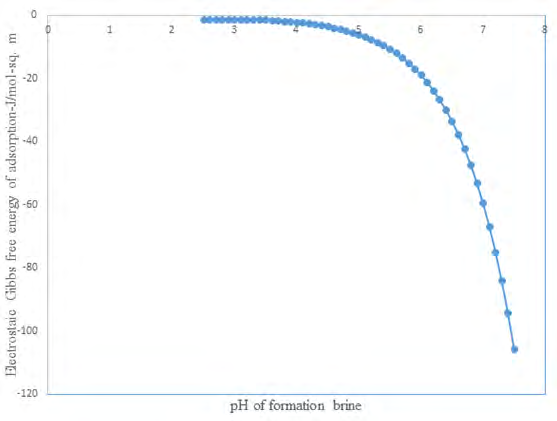
Discussion
Implication of Gibbs Free Energy Trend for Wettability Evolution
In this paper the free energy given by Equation 23 which was derived using equation 7 will be assumed to be related to ion adsorption at the silica-brine interface in geologic systems. Therefore, the free energy will be equated to adsorption free energy. In this regard, Figure 1 shows a theoretical plot of Gibbs free energy variation with pH of formation brine that will be typically encountered when carbon dioxide is injected into sandstone saline aquifers. Values of free energy of adsorption calculated using Equation 23 are negative. Elsewhere Malin, et al. [34] (appendix 1), the free energy of adsorption at the silica-water interface has been studied and values of adsorption free energy have the same sign. The range of pH was chosen because dissolution of carbon dioxide in brine will produce increase acidity conditions that will cause pH reduction of formation brine [28]. Apparently, Gibbs free energy increases with decreasing pH reduction and attains a maximum value at pH of formation brine equal to 3 which is the point of zero charge pH of silica surface. To discuss the implication for wettability evolution two notable theories in the colloidal science will be invoked. They are intermolecular forces theory and disjoining pressure forces theory [35].
In line with intermolecular forces theory, the interfacial tension between solid and liquid is given by Equation 5. This is recalled below:
2 1/2 1/2 ( ) ( )
= − + + − −
γ γ γ LW LW ij i j ( ) ( ) ( ) ( )
1/2 1/2 1/2 1/2 2 γ γ γ γ γ γ γ γ + − + − + − − +
i i j j i i i j Under geological sequestration conditions pertaining to sandstones saline aquifers, the surface charge which corresponds to electrostatic contribution to solid-liquid interfacial tension will be predominantly negative, meaning zero surface positive charge. Consequently, this equation can be written as:
2 1/2 1/2 1/2 1/2 LW LW ij i J j j j j γ γ γ γ γ γ γ + − − + = − + + − ( ) ( ) ( ) ( ) Thermodynamically, as Gibbs free energy increases following pH decreases, surface charging reaction which is normally described by Equation 2 which is deprotonation of silanol surface species at high pH conditions will reduce. Therefore, surface charges are reduced with pH decreases with reaction 1 being favored which is protonation in nature. The surface of silica will be gradually approaching a state of zero charge and will attain this when the pH of formation brine has reduced to 3, which is the point of zero charge pH of silica surface. In relationship to the above modified equation this means that negative surface species are depleting due to deprotonation reaction in response to pH decreases which accounts for increase hydrogen ion activity of formation brine. Consequently, this equation can be interpreted to mean that as negative surface charges decrease the electrostatic contribution to solid-liquid interfacial tension will decrease. The mathematical interpretation is that solid- liquid interfacial tension will increase with decreasing pH. Accordingly, it will attain a maximum at the point of zero charge pH when the surface charge of silica is zero. Therefore, the implication for wettability evolution can be appreciated by invoking Young’s equation for wettability given by Equation 24 assuming constant liquid-vapor and solid-vapor interfacial tensions [36]:
γ γ − = (24) cos lv sl γ lv Accordingly, as solid-liquid interfacial tension increases, following pH decreases the numerator of this equation is decreasing. Therefore, the cosine of the macroscopic contact angle, which is quantitative measure of wettability, will decrease. In this regard, minimum wettability will occur when the surface charge is zero, corresponding to maximum point of Gibbs free energy as seen the (Figure 1).
The analogy of this pH induced wettability reduction can be seen in electro wetting theory where Lippmann understood the metal-electrolyte interfacial tension to evolve in accordance with the following equation [37]:
γ σ = − (25) sl d dV Where sl γ is solid-liquid interfacial tension, V is the voltage applied across the interface and σ is the surface charge density.
This equation leads to the following:
sl slo dV γ γ σ = − (26)
Where, slo γ , is the solid-liquid interfacial tension neglecting an electrical voltage.
This equation means that when there is an applied voltage, there is decrease in solid-liquid interfacial tension. Consequently, as the applied voltage decreases solid-liquid interfacial tension increases, corresponding to decreases in surface charge density as encountered in geologic systems with wettability reduction similar to that encountered in geologic systems.
In the electric double layer theory, disjoining pressure forces are those that tend to separate two interfaces [38]. This force has three fundamental components. They consist of van de Waals electrodynamic forces which are attractive in origin, double layer repulsive forces [39] which are repulsive and short range structural and salvation forces [40] which are also repulsive in origin. Structural or solvation components result from the structuring of water molecules [41] close to solid surface which changes the dielectric permittivity leading to violation of the classical DVLO (Boris Derjaguin and Lev Landau, Evert Verwey and Theodoor Overbeek) theory [42] which assumes dielectric continuum [43]. Theoretically, double layer repulsive forces originate from electrostatic phenomena related to surface electrical charges on solids as encountered in geologic and colloidal systems. Since they are electrical in nature, their repulsive nature which stabilizes the thin wetting film on solids will be proportional to the magnitudes of surface charges. In the context of the present paper, as surface charges decrease due to pH decreases and as Gibbs free energy increases, there will be reduction of disjoining pressure forces due double layer repulsion. Therefore, the interface will tend to approach each other and this causes destabilization of the thin wetting film leading to dewetting phenomena [35]. This arises because of competition between attractive van der Waals attractive forces and repulsive forces furnished by the other two components of disjoining pressure [44]. Furthermore, in the context of the present paper, pH decrease and Gibbs free energy increase correspond to decrease in double layer repulsion to cause destabilization of the thin wetting films in geologic systems since this force is responsible for enhanced thin film stability [45].
Relationship of Present work to Published work in the Literature
The theoretical justification of our pH dependent Gibbs free energy of the electric double layer is seen in the context of recent trends in contact angle for the carbon dioxide- brine solid systems. In this regard, Kim, et al. [46] studied experimentally the wettability of the system carbon dioxide- silica-brine. They concluded from their experimental findings that interactions of supercritical carbon dioxide with brines in the presence of silica surface reduce wettability. Jung and Wan [47], also studied this system experimentally under supercritical gas conditions. By using two independent approaches for contact angle measurements, their summary conclusion is that interaction of supercritical carbon dioxide with reservoir rock in the presence of brine increased contact angle which corresponds to wettability decreases similar to that reported by Kim, et al. [46]. Similar observations have also been reported elsewhere [48]. To determine the effect of supercritical carbon dioxide interaction on the capillary sealing capability of shale cap rocks over potential saline aquifers [49], used experimental approaches. They arrived at a similar conclusion. Therefore, in the context of the present paper, carbon dioxide injection into sandstone saline aquifers and the resulting decrease in formation brine pH corresponds to increases in Gibbs free energy of electrostatic interaction and this has the effect of wettability decreases as explained theoretically using the outcome of the final equation in this paper. In the work of Malin, et al. [34] the adsorption free energy at the silica-water interface is plotted as a function of initial surface potential (Appendix 1). In the mentioned paper, as initial surface potential changes from high negative to low negative, the free energy of adsorption increases from a high negative value to a low negative value. Apparently, low negative values of initial surface potential correspond to low pH of aqueous solution and since this leads to increases in free energy of adsorption, values calculated in the present work are realistic, considering the silica-water interface.
Implication for Efficient Carbon Storage in Saline Aquifers
In the theory of multiphase flow in porous media, the concept of wettability and its intimate link to relative mobility of fluid phases abounds in the petroleum literature Owens & Archer [50]; Anderson [51]; Chilingara & Yena [52]. Generally, the more wetting a phase is with considering the solid surface, the lower is its relative mobility compared to the non-wetting fluid. Consequently, if the injection of carbon dioxide into a saline aquifer leads to decreases in wettability of the aquifer rock considering resident brine, its relative mobility will be higher compared to that of injected carbon dioxide. Consequently, injected carbon dioxide will move behind displaced formation brine with a reduced relative mobility, which enhances favorable mobility ratio and sweep efficiency in geological carbon storage. Moreover, putting the experimental findings of Kim and Wang [46] and Jung and Wang [47] where contact angle increases with pressure in the context of our theoretical model, it suffices to say that as carbon dioxide pressure increases, increased dissolution of the gas will induce more acidity and lower pH of aqueous solution. The implication is that the Gibbs free energy of the electric double layer which is linked to the electrostatic free energy of surface charging will increase and that leads to low surface charge density/potential which increases solid- liquid interfacial tension, leading to contact angle increase in accordance with Young’s equation.
Conclusion
Adsorption phenomena at the silica-water interface in geologic systems is intimately linked to electrical phenomena. These electrical phenomena are directly related to the electrical double layer at the interface. Based on the molecular theory of interfacial tension, electrical phenomenon at the solid-liquid interface will impact interfacial tension. Therefore, for a multiphase flow regime specified by carbon dioxide and water in porous media, electrostatic effect on solid-liquid tensions will impact wettability. This process has to do with electrical surface charging which can be linked to the free energy of adsorption at silica-water interface. This paper has used an original expression for the free energy of the electrical double layer to derive a pH dependent adsorption free energy for the silica- water system under geologic carbon storage. The following sum up the conclusions of this paper:
- The pH dependence of the free energy of the electric double layer can be described by a hyperbolic equation.
- Increasing Gibbs free energy of the electric double layer will occur until a maximum corresponding to zero occurs at the point of zero charge pH corresponding to a maximum value of the solid-liquid interfacial tension.
- The pH dependent Gibbs free energy equation in this paper explains increases in free energy which has a direct link to increase solid-liquid interfacial tension as pH decrease and can, therefore, be used to explain recent experimental trends on contacts for the carbon dioxide- brine-solid systems.
Acknowledgements
We wish to acknowledge the timely contributions to our work by the document delivery section of the Library, and the Office of Research and Graduate Studies, Cape Breton University. They have made it possible to complete this manuscript on time.
References
-
Tsuneo I, Mizutani H (1981) Experimental and theoretical basis of electrokinetic phenomena in rock- water systems and its applications to geophysics. Journal of Geophysical Research 86(B3): 1763-1775.
-
Barger JR, Parks GA, Brown GE (1997) Surface complexation of Pb(II) at oxide-water interfaces: II. XAFS and bond-valence determination of mononuclear Pb(II) sorption products and surface functional groups on iron oxides. Geochimica et Cosmochimica Acta 63(13): 2639- 2652.
-
Sverjensky DA (1994) Zero-point-of-charge prediction from crystal chemistry and solvation theory. Geochimica et Cosmochimica Acta 58(14): 3123-3129.
-
Revil A, Schwaegear H, Cathles L, Manhardt PD (1999) Streaming potential in porous media: 2. Theory and application to geothermal systems. Journal of Geophysical Research 104(B9): 20033-20048.
-
Holness MB (1992) Equilibrium dihedral angles in the system quartz-CO2 single bond H2O single bond NaCl at 800°C and 1–15 kbar: the effects of pressure and fluid composition on the permeability of quartzites. Earth and Planetary Science Letters 114(1): 171-184.
-
Kollman P (1993) Free energy calculations: Applications to chemical and biochemical phenomena. Chem Rev 93(7): 2395-2417.
-
Dunwell M, Yan Y, Xu B (2018) Understanding the influence of the electrochemical double-layer on heterogeneous electrochemical reactions. Current Opinion in Chemical Engineering 20: 151-158.
-
Amadu M, Midonye A (2019) Derivation of a pH Dependent Solid-Liquid Interfacial Tension and Theoretical Interpretation of the Physicochemistry of Dewetting in the CO2-Brine-Silica System. International Journal of Chemistry 11(2): 127-155
-
Předota M, Machesky ML, Wesolowski DJ, Cummings PT (2013) Electric Double Layer at the Rutile (110) Surface. 4. Effect of Temperature and pH on the Adsorption and Dynamics of Ions. J Phys Chem C 117(44): 22852-22866.
-
Fokkink LGJ, Keizer AD, Lyklema J (1989) Temperature Dependence of the Electric Double Layer-on Oxides: Rutile and Hematite. Journal of Colloid and Interface Science 127(1): 116-131.
-
Yamaguchi Y, Kusudo H, Surblys D, Omori T, Kikugawa G (2019) Interpretation of Young’s equation for a liquid droplet on a flat and smooth solid surface: Mechanical and thermodynamic routes with a simple Lennard-Jones liquid. J Chem Phys 152: 179901.
-
Dugger DL, Stanton JH, Irby BN, McConnell BL, Cummings WW, et al. (1964) The Exchange of Twenty Metal Ions with the Weakly Acidic Silanol Group of Silica Gel J Phys Chem 68(4): 757-760.
-
Zhuravlev LT, Potapov VV (2006) Density of Silanol Groups on the Surface of Silica Precipitated from a Hydrothermal Solution. Russian Journal of Physical Chemistry 80(7): 1119-1128.
-
Glover PW, Meredith PG, Sammonds PR, Murrell SA (1994) Ionic surface electrical conductivity in sandstone. Journal of Geophysical Research 99(B11): 21635-21650.
-
Fowkes FM (1964) Attractive Forces at Interfaces. Ind Eng Chem 56(12): 40-52.
-
Saito M, Yabe A (1984) Dispersion and Polar Force Components of Surface Tension of Oily Soils. Textile Research Journal 54(1): 18-22.
-
Oss CJ, Good RJ, Chaudhury MK (1988) Additive and nonadditive surface tension components and the interpretation of contact angles. Langmuir 4(4): 884- 891.
-
Atkinson RJ, Posner AM, Quirk JP (1967) Adsorption of potential-determining ions at the ferric oxide-aqueous electrolyte interface. J Phys Chem 71(3): 550-558.
-
Hiemstra T, Riemsjik WH (1996) A Surface Structural Approach to Ion Adsorption: The Charge Distribution (CD) Model. Journal of Colloid and Interface Science 179(2): 488-508.
-
Xiao L, Honig B (1999) Electrostatic Contributions to the Stability of Hyperthermophilic Proteins. J Mol Biol 289(5): 1435-1444.
-
Wirth MJ, Fairbank RW, Fatunmbi HO (1997) Mixed Self- Assembled Monolayers in Chemical Separations. Science 275(5296): 44-47.
-
Healy TW, White LR (1978) Ionizable surface group models of aqueous interfaces. Advances in Colloid and Interface Science 9(4): 303-345.
-
Revil A, Pezard PA (1999) Streaming potential in porous media: 1. Theory of the zeta potential. Journal of Geophysical Research 104(B9): 20021-20031.
-
Johnson SB, Drummond CJ, Scales PJ, Nishimura S (1995) Comparison of Techniques for Measuring the Electric Double Layer Properties of Surfaces in Aqueous solutions: Hexadecytrimethylammonium Bromide Self- Assembly Structures as a Model. Langmuir 11(7): 2367- 2375.
-
Ishido T, Mizutani H (1981) Experimental and Theoretical Basis of Electrokinetic Phenomenain Rock- Water Systems and Its Applications to Geophysics. Journal of Geophysical Research 86(B3): 1763-1775.
-
Simonson BM (1987) Early Silica Cementation and Subsequent Diagenesis in Arenites from Four Early Proterozoic Iron Formations of North America. Journal of Sedimentary Research (SEPM) 57(3): 494-511.
-
Kosmulski M (2006) pH-dependent surface charging and points of zero charge III Update. Journal of Colloid and Interface Science 298(1): 730-741.
-
Benson SM, Cole DR (2008) CO2 Sequestration in Deep Sedimentary Formations. Elements 4(5): 325-331.
-
Vainrub A, Pettitt BM (2000) Thermodynamics of association to a molecule immobilized in an electric double layer. Chemical Physics Letters 323(1-2): 160- 166.
-
Hanor JS (1994) Origin of saline fluids in sedimentary basins. Geological Society, London, Special Publications 78: 151-174.
-
Stogryn A (1971) Equations for Calculating the Dielectric Constant of Saline Water (Correspondence). Microwave Theory and Techniqu 19(8): 773-736.
-
Malmber C, Maryott A (1956) Dielectric Constant of Water from 0 to 100 Degrees Celsius. Journal of Research of the National Bureau of Standard 56(1): 1-6.
-
Childs WHJ (1972) Physical Constants. Springer.
-
Malin JN, Holland JG, Geiger FM (2009) Free Energy Relationships in the Electric Double Layer and Alkali Earth Speciation at the. J Phys Chem 113(41): 17795- 17802.
-
Basu S, Sharma MM (1996) Measurement of Critical Disjoining Pressure for Dewetting of Solid Surfaces. Journal of Colloid and Interface Science 181(2): 443-455.
-
Roura P, Fort J (2004) Local thermodynamic derivation of Young’s equation. Journal of Colloid and Interface Science 272(2): 420-429.
-
Quilliet C, Berge B (2001) Electrowetting: a recent outbreak. Current Opinion in Colloid & Interface Science 6(1): 34-39.
-
Hirasaki G (1991) Wettability: Fundamentals and Surface Forces. SPE Formation Evaluation 6(2): 217-226.
-
Miklavi SJ, Chan DY, White LR, Healy TW (1994) Double Layer Forces between Heterogeneous Charged Surfaces. J Phys Chem 98(36): 9022-9032.
-
Churaev ND, Derjaguin BV (1985) Inclusion of structural forces in the theory of stability of colloids and films. Journal of Colloid and Interface Science 103(2): 542-553.
-
Israelachvili J (1987) Solvation forces and liquid structure, as probed by direct force measurements. Acc Chem Res 20(11): 415-421.
-
Ninham B (1999) On progress in forces since the DLVO theory. Advances in Colloid and Interface Science 83(1- 3): 1-17.
-
Kjellander R, Marcelja S, Pashley RM, Quirk JP (1988) Double-layer ion correlation forces restrict calcium-clay swelling. J Phys Chem 92(23): 6489-6492.
-
Senden TJ, Drummond CJ, Kekicheff P (1994) Atomic Force Microscopy: Imaging with Electrical Double Layer Interactions. Langmuir 10(2): 358-362.
-
Reiter G, Ashutosh S, Alain C, Odile DM, Rajesh K, et al. (1999) Thin Film Instability Induced by Long-Range Forces. Langmuir 15(7): 2551-2558.
-
Kim Y, Wan J, Kneafsey JT, Tetsu KT (2012) Dewetting of Silica Surfaces upon Reactions with Supercritical CO2 and Brine: Pore-Scale Studies in Micromodels. Environ Sci Technol 46(7): 4228-4235.
-
Jung JW, Wan J (2012) Supercritical CO2 and Ionic Strength Effects on Wettability of Silica Surfaces: Equilibrium Contact Angle Measurements. Energy Fuels 26(9): 6053-6059.
-
Dickson JL, Gupta G, Horozov TS, Binks BP, Johnston KP (2006) Wetting Phenomena at the CO2/Water/Glass Interface. Langmuir 22(5): 2161-2170.
-
Chiquet PD, Broseta D, Thibeau S (2007) Wettability alteration of caprock minerals by carbon dioxide. Geofluids 7(2): 112-122.
-
Owens W, Archer D (1971) The Effect of Rock Wettability on Oil-Water Relative Permeability Relationships. J Pet Technol 23(7): 873-875.
-
Anderson WG (1987) Wettability Literature Survey Part 5: The Effects of Wettability on Relative Permeability. J Pet Technol 39(11): 1453-1468.
-
Chilingara GV, Yena TF (1983) Some Notes on Wettability and Relative Permeabilities of Carbonate Reservoir Rocks, II. Energy Sources 7(1): 67-75.
- Nigeria’s Vulnerability in the Face of Global Energy Policy
- A Simulation Study of Investigation of Optimum Oil Production Performance by Applying Various Gas Injection Methods in Oil Reservoir
- Characterization of Permo-Triassic Reservoirs through Thermal Maturity Assessment of Westphalian Source Rocks in the Cheshire Basin
- Influence of Microwax on the Rheological and Thermal Behaviour of a Wax Crude Oil
- Real-Time Monitoring and Performance Optimization of Steam Injection in Heavy Oil Reservoirs Using Fiber Optic Sensing and Integrated Predictive Simulation Models
- Rapid On-Site Determination of the Total Petroleum Hydrocarbon Content of Soils by Handheld Fourier Transform Near-Infrared Spectroscopy: Development of a Global, Site- and Scanner- Independent Calibration Model