Amyloidosis: An Act of Deception in the Liver
We report a case of end-stage liver disease initially attributed to acute on chronic liver failure (ACLF) in a 59 YO African American female. She presented encephalopathic with stable vital signs. Her laboratory tests were as follows: bilirubin: 24.3 mg/dL, INR 2, Creatinine 4.2 mg/dL, MELD of 40 with rapidly declining renal function. Magnetic resonance cholangiopancreatography (MRCP) was negative. Her serologic work-up was unrevealing. She was deemed to have decompensated cryptogenic cirrhosis for which she received simultaneous liver & kidney transplantation. Her explanted liver revealed extensive amyloidosis, AL Amyloid kappa type. Urine electrophoresis showed a monoclonal free kappa light chain. Bone marrow biopsy and flow cytometry were negative for plasma cell dyscrasia. Further work up suggested no systemic involvement. We present this rare case of primary hepatic amyloidosis and discuss the importance of this diagnostic consideration in patients with otherwise unclear etiology of liver disease.
Introduction
Amyloidosis is a rare disorder where the improper deposition of an insoluble protein in tissue which impairs its function and architecture, leading to organ failure and death [1]. Hepatic involvement in the setting of systemic amyloidosis is a common occurrence, seen in 62-90% of patients, with manifestations limited to hepatomegaly and elevated alkaline phosphatase levels. Stigmata of chronic liver disease are rare: hyperbilirubinemia is seen in 5% of patients and suggests a poor prognosis. Our patient presented with hyperbilirubinemia, jaundice, and weight loss without hepatomegaly or evidence of systemic amyloidosis. Her condition progressed to acute on chronic liver failure (ACLF) and renal failure without a clear etiology requiring simultaneous liver-kidney transplantation. Her explanted liver showed massive amyloid deposition in the portal and periportal regions-a topographical distribution that is extremely rare. Manifestations of hepatic amyloidosis are usually limited to hepatomegaly and abnormal lab values but can present as hepatic failure and should be a diagnostic consideration in patients with otherwise unclear etiology of liver disease.
We describe a case of a 57-year-old African American female with a history of multiple sclerosis, hypertension and gastroesophageal reflux disease that presented to our hospital for evaluation of persistent hyperbilirubinemia and elevated hepatic enzymes.
While previously in good health, she presented to an outside hospital after a fall and was incidentally noted to have elevated transaminases. This transaminase elevation was thought to be due to cholecystitis. The patient subsequently underwent a cholecystectomy without any improvement in liver enzymes. Post-operatively, there was a complication of a blood clot in the bile duct which required an endoscopic retrograde cholangiopancreatography (ERCP) and stent placement. However, the patient’s transaminases and bilirubin remained persistently elevated. A repeat ERCP was performed which showed biliary sludge in the bile duct and the stent was removed as the bile duct and ampulla were patent and unobstructed. The patient was then started on steroid therapy.
Despite these therapies, the patient’s transaminases and bilirubin levels remained elevated and showed no significant improvement. The patient started to report symptoms of jaundice, scleral icterus, diffuse pruritus and an approximate 20 lbs weight loss over several months. Pertinent physical exam findings revealed jaundice, scleral icterus, and a soft non-distended abdomen without ascites. Hepatosplenomegaly was not appreciated on physical examination. Laboratory studies at initial presentation to our hospital revealed a total bilirubin of 18.4 mg/dL, direct bilirubin 13.6 mg/dL, alkaline phosphatase 267 U/L, ALT 53 U/L, and AST 66 U/L. Viral, AMA, ASMA, A1AT, copper and iron serologies were negative.
The working differential diagnosis at the time included drug-induced liver injury (DILI), however there was no clear causative medication, and concern for extrahepatic biliary obstruction such as primary sclerosing cholangitis and less likely primary biliary cholangitis.
The prior outside liver core needle biopsy was reviewed and showed cholestasis with ductular reaction and associated portal fibrosis and portal inflammation. In addition, there were frequent foci of lobular inflammation. The native bile ducts appeared intact without overt bile duct injury noted. Significant ductular reaction was noted in many of the portal tracts, with bile plugs in some of the proliferating ductules. The provided trichrome stain showed normal liver parenchymal architecture with an initial interpretation of portal fibrosis. Because of this apparent cholestatic pattern, this gave the overall suggestion of extrahepatic biliary obstruction, including primary sclerosing cholangitis (PSC). Drug induced liver injury (DILI) was also a consideration, prompting discontinuation of a hypertensive medication (Lisinopril) but was considered less likely considering her 20 year history of taking the medication.
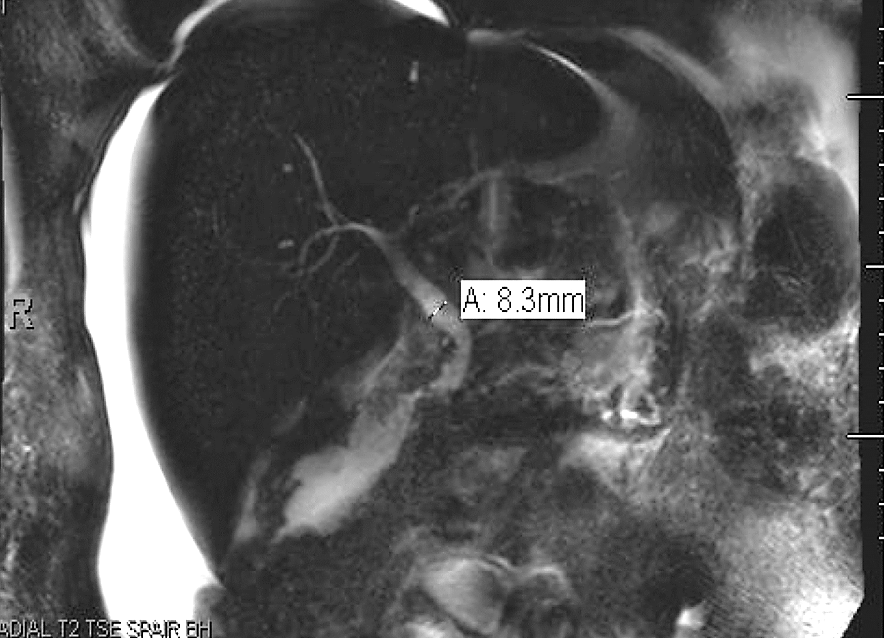
Magnetic resonance cholangiopancreatography (MRCP) was subsequently performed which revealed a mildly enlarged liver with no intrahepatic biliary duct dilation. However, there was mild dilatation of the common bile duct with smooth tapering and without evidence of choledocholithiasis or obstructing lesions (Figure 1). There was no abnormal beading of the intrahepatic bile ducts to suggest PSC. No focal hepatic masses were identified, and there was moderate to large amount of ascites.
The patient continued to deteriorate with persistent hyperbilirubinemia, weight loss, intermittent confusion and ascites. The patient had started to require large volume paracentesis every 1-2 weeks. In addition, ursodiol, lasix and spironolactone were added to her therapeutic regimen. Despite continued therapies, the patient’s clinical picture of cholangitis persisted and developed into decompensated cryptogenic cirrhosis. The patient’s MELD scores had increased from 19 at initial evaluation to 25 a year later. The patient was then worked up for transplant evaluation.
During workup for hepatic transplant, the patient was admitted to an OSH for decompensated cryptogenic cirrhosis, hepatic encephalopathy and acute on chronic kidney failure and transferred to our institution for further evaluation and treatment. The patient’s renal failure was presumed to be secondary to hepatorenal syndrome with a baseline Creatinine of 1.5 mg/dl was elevated to 4.5 on presentation. The patient was managed with lactulose, octreotide and misoprostol, as well as broad spectrum antibiotics. She was started on continuous renal replacement therapy (CRRT). Despite aggressive treatment, the patient’s renal function had continued to decline and she met criteria for kidney transplantation as well.
Due to the patients acute on chronic liver failure from cryptogenic cirrhosis and acute on chronic kidney failure, the patient was listed for simultaneous liver/kidney transplantation, MELD-Na score of 43. After a donor was found, the patient underwent liver and kidney transplantation with the explanted liver was sent for histopathologic examination. The native kidney was not removed during this procedure, thus was not examined histologically.
On macroscopic examination, the liver weighed 1.5 kg and showed a granulated brown-tan external surface and yellow- green waxy material covering the capsule. The parenchyma was brown-tan, granular and without an identifiable mass.
Microscopic evaluation revealed evolving hepatic cirrhosis with extensive amyloid deposition in the portal and periportal areas. This was confirmed on a Congo red stain which showed the characteristic apple-green birefringence on polarized light. Some of the amyloid was seen extending into the fibrous septae (Figure 2). The paraffin-embedded block was evaluated by liquid chromatography tandem mass spectrometry (LC MS/MS) for typing which revealed a peptide profile consistent with an AL (Kappa-type) amyloid deposition.
At this time, the workup for amyloidosis was pursued and confirmed, which provided no evidence of involvement of another organ, but did provide other laboratory values consistent with the disease. Upon recognition of this pathology, electrophoresis studies revealed a poorly defined IgA band in the serum and a monoclonal free kappa light chain in the urine. A subsequent bone marrow biopsy failed to reveal either plasma cell dyscrasia or amyloid deposition. Skeletal survey was without any suspicious lytic lesions. Echocardiogram showed left ventricular hypertrophy and an elevated BNP. Urine protein immunofixation showed monoclonal light chains in the urine. The patient was then sent to her local oncologist for follow up and management.
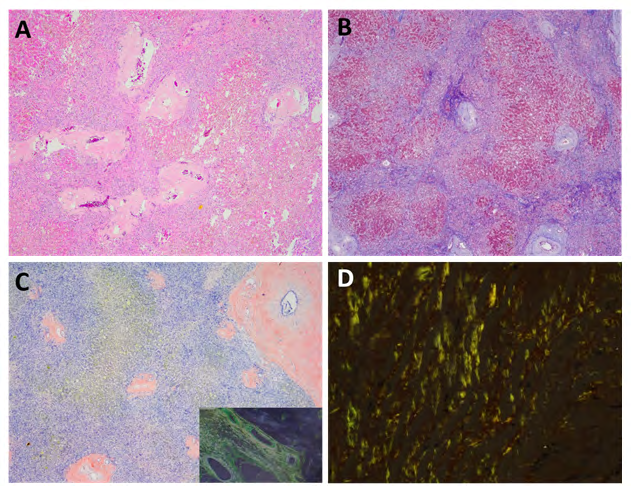
Figure 2: Explant liver histologic studies. A: Hematoxylin and Eosin stain shows deposition of amorphous material within the portal areas consistent with amyloid. B: Trichrome stain confirms the cirrhotic nature of the liver. C: Congo ted stain highlights the amyloid deposition, which shows apple-green birefringence under polarized light (inset). D: High power view of the amyloid birefringence under polarized light.
Discussion
Amyloidosis is a relatively uncommon disease characterized by deposition of insoluble fibrillar fragments of monoclonal light chains. Park MA, et al. [2] these protein fragments are produced by a monoclonal population of plasma cells. These insoluble protein fragments subsequently enter the circulatory system and deposit into multiple organs causing dysfunction. Though not as frequent as heart or kidney involvement, the liver is frequently affected; one study showed that 70% of cases of primary systemic amyloidosis had liver involvement. Hepatic infiltration although common, is rarely of clinical importance [3]. Notably, non-specific symptoms such as involuntary weight loss, fatigue, abdominal pain, edema and anorexia are found in these patients. Physical examination findings in hepatic amyloidosis patients are also nonspecific and include hepatomegaly, ascites, and purpura, Park MA, et al. [2] none of which were exhibited by our patient on initial presentation. Common laboratory findings include elevated alkaline phosphatase, aspartate aminotransferase (AST), and total cholesterol. Park MA, et al. [2] Proteinuria has found to be present in 90% of patients with hepatic amyloidosis in several studies with total serum bilirubin levels are only elevated in a minority of patients [4].
In our case, the patient presented with nonspecific clinical features including decompensated liver failure, hepatic encephalopathy, hepatomegaly, and elevated bilirubin. Our patient had documented weight loss of 20 pounds and had complained of diffuse pruritus, jaundice, and scleral icterus 6 months prior to presentation with persistently elevated transaminases and bilirubin. But as you can see, the laboratory findings are incredibly nonspecific and even if amyloidosis was suspected, tissue diagnosis is needed for confirmation with a liver biopsy remaining the definitive method for confirmation of hepatic involvement by amyloidosis. In addition, this case represents isolated hepatic involvement of amyloidosis as no evidence of extrahepatic involvement was noted. In addition, amyloid deposition may show positive staining expression on trichrome stain, mimicking portal fibrosis. This was depicted on retrospective review of the prior needle core biopsy, which highlights amyloid deposition on Congo red stain.
Typically, the histopathology of Hepatic amyloidosis is characterized by the presence of sinusoidal deposits of amyloid within the spaces of disse or deposits within the walls of blood vessels [5, 6]. From Sarsik, et al. [7] a pure sinusoidal or pure vascular pattern of amyloid deposition is seen in 70% of specimens, with the pure vascular patterns representing ~65% and the pure sinusoidal pattern representing ~6%. Sarsik, et al. [7] in that same study, the portal stromal pattern was seen in one patient with a mixed vascular deposition pattern. Therefore, the pure portal stromal pattern which is seen in isolation in our case has not been described in their study. In a study by authored by Iwata, et al, there is noted that some AL amyloidosis show interstitial involvement of the portal triad, without vessel or bile duct involvement [8]. However, of the 117 AL-type Hepatic amyloidosis cases reviewed in that study, only 6 (~5%) showed the pure portal stromal pattern we describe in our case [8].
Conclusion
We present this rare case of primary hepatic amyloidosis showing a deceptive and inconspicuous portal tract stromal deposition pattern. This deserves diagnostic consideration in cryptogenic cases of liver dysfunction of unclear etiology. Isolated deposition of amyloid into the portal stroma poses a diagnostic challenge on liver core biopsy as it is an unusual pattern of deposition which can mimic portal fibrosis on routine H&E slides. In addition, this deception can be compounded by positive staining for trichrome, which can easily be interpreted as portal fibrosis.
As was the case with our patient, positive trichrome staining of this area alone can lead to the impression of portal fibrosis. The clinical sequelae of high grade bile outflow obstruction further confounded the diagnosis, as entities which can cause portal fibrosis remained in the differential diagnosis. It was not until histopathologic analysis of the explanted liver where hepatic amyloidosis was diagnosed. This was important as the explant provided much more tissue surface area than the biopsy and thus more portal tracts for evaluation.
Still, the broader implication of this case is that of a diagnostic pitfall imposed by the very rare pattern of isolated portal tract stromal amyloidosis. In the case of the patient with liver failure of unclear etiology, amyloidosis should remain a consideration.
References
-
Baker KR, Rice L (2012) The amyloidoses: clinical features, diagnosis and treatment. Methodist Debakey Cardiovasc J 8(3): 3-7.
-
Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, et al. (2003) Primary (AL) hepatic amyloidosis: clinical features and natural history in 98 patients. Medicine (Baltimore) 82(5): 291-298.
-
Buck FS, Koss MN (1991) Hepatic amyloidosis: morphologic differences between systemic AL and AA types. Hum Pathol 22(9): 904-907.
-
Gertz MA, Kyle RA (1988) Hepatic amyloidosis (primary [AL], immunoglobulin light chain): the natural history in 80 patients. Am J Med 85(1): 73-80.
-
Sonthalia N, Jain S, Pawar S, Zanwar V, Surude R, et al. (2016) Primary hepatic amyloidosis: A case report and review of literature. World J Hepatol 8(6): 340-344.
-
Wang YD, Zhao CY, Yin HZ (2012) Primary hepatic amyloidosis: a mini literature review and five cases report. Annals of Hepatology 11(5): 721-727.
-
Sarsik B, Sen S, Kirdok FS, Akarca US, Toz H, et al. (2012) Hepatic amyloidosis: morphologic spectrum of histopathological changes in AA and nonAA amyloidosis. Pathol Res Pract 208(12): 713-718.
-
Iwata T, Hoshii Y, Kawano H, Gondo T, Takahashi M, et al. (1995) Hepatic amyloidosis in Japan: histological and morphometric analysis based on amyloid proteins. Hum Pathol 26(10): 1148-1153.
- Management of Gallbladder Perforations: A Review
- From The Mouth to the Gut: The Oral Microbiome's Role in Promoting Gastrointestinal Disease
- Case Report: Intraductal Papillary Mucinous Neoplasm (IPMN) Complicated by Portal Vein Plaquing and Biliary Obstruction Mimicking Pancreatic Metastatic Malignancy
- Management of Non-Cirrhotic Portal Hypertension during Pregnancy: A Review
- Effectiveness of Omeprazole versus Pantoprazole for Symptomatic Relief of Gastro-Esophageal Reflux Disease (GERD)/ Acid Peptic Disease (APD): A Real-World Evidence (RWE) Study
- Case of Splenic Infarction; A Rare Presentation of Complicated Enteric Fever in a Pediatric Patient