Insight into the Pathophysiology of Polycystic Ovarian Syndrome: The Genes are not to Blame?
Polycystic ovarian syndrome (PCOS) is a common and complicated endocrine disorder affecting women of reproductive age group mainly. The characteristics features include an ovulatory menstrual changes, hyperandrogenism and polycystic ovaries. It is diagnosed using various diagnostic criteria but the Rotterdam criteria are the most commonly referenced. PCOS is associated with cardiovascular risk factors such as glucose intolerance, hypertension, obesity and dyslipidaemia. Noncardiovascular complications such as subfertility, endometrial cancer, obstetric complications and psychological disturbance are also significant. The pathophysiology arises from the interactions between the predisposing genes and the environment. The pathophysiologic mechanisms of PCOS such as insulin resistance, altered androgen secretion and action, dysregulated gonadotropin action and chronic subclinical inflammation have genetic bases. The role of genes in the pathophysiology of PCOS is still an evolving concept and efforts are ongoing in gaining further understanding about the disease despite the challenges of genetic studies.
Introduction
Polycystic ovarian syndrome (PCOS) is a common endocrine disorder in young women characterized by clinical or biochemical hyperandrogenism and chronic an ovulatory menstrual changes [1]. PCOS is a heterogeneous constellation of clinical features that form a spectrum of disorders. At one end of the spectrum are patients with mild presentation and at the other end are patients presenting with severe disturbance of reproductive, endocrine and metabolic function. There are multiple criteria used in making the diagnosis but the most commonly referenced is the Rotterdam criteria. PCOS is a disorder that adversely affects the homeostasis of the tightly regulated hypothalamic-pituitary-ovarian endocrine axis. It affects 5-20 % of women of reproductive age group making it the most common endocrine disorder in this age group [2]. The most commonly reported clinical features are hirsutism, oligomenorrhoea/amenorrhoea, infertility and obesity.
PCOS is associated with a higher incidence of traditional cardiovascular risk factors such as glucose intolerance (including gestational diabetes mellitus), hypertension, obesity and dyslipidaemia [3]. In addition, higher incidence of non-cardiovascular diseases such as endometrial hyperplasia/cancer and mood disorders have been reported in patients diagnosed with PCOS. Other obstetric complications such as miscarriage, pre-eclampsia, preterm delivery and increased Caesarean sections have been documented in PCOS.
Among the cardiovascular risk factors that PCOS is predisposed to, type 2 diabetes occupies a central place. Diabetes mellitus is a heterogeneous group of metabolic disorders characterized by chronic hyperglycaemia due to a defect in insulin secretion and/or action. According to the most recent classification of diabetes by the World Health Organization (WHO), diabetes is grouped into type 1diabetes, type 2 diabetes, hybrid type, specific types, hyperglycaemia first detected in pregnancy and the unclassified type [4]. Type 2 diabetes is the commonest type of diabetes and it is the one often developed by patients with PCOS.
Fifty percent of individuals with PCOS will develop type 2 diabetes within 10 years [5]. PCOS is considered an independent risk factor for developing type 2 diabetes and from progressing from impaired glucose tolerance to type 2 diabetes. A major co-founder in the association between PCOS and type 2 diabetes is obesity which is present in both conditions. In fact, it is estimated that patients with PCOS and obesity are three to four times more likely to develop type 2 diabetes compared with those with PCOS but without diabetes [6]. Chronic sub-clinical inflammation has been the main hypothesis to explain the association between PCOS and cardiovascular diseases. This has been confirmed in multiple studies which have reported a higher level of C-reactive protein (a marker of inflammation) in patients with PCOS compared with the general population. In addition, type 2 diabetes occurs much earlier with PCOS compared with women without PCOS. Consequently, women with PCOS are screened earlier and more frequently for type 2 diabetes compared with the general population. Central to the pathophysiology of type 2 diabetes is insulin resistance. Insulin resistance is defined as the attenuated response of tissues to a given amount of insulin. As a result, the body has to produce a higher amount of insulin to get the same response. With time, there is exhaustion of the physiological reserve of β- cells of the pancreatic islets of Langerhans hence, type 2 diabetes develops.
Insulin resistance is considered as the bridge between PCOS and type 2 diabetes. All the other co-founders such as obesity, hyperandrogenism, hypertension and dyslipidaemia are all linked to insulin resistance. Studies have shown that when these co-founders are treated, insulin sensitivity improves remarkably [7]. Some researchers have proposed muscle mitochondria dysfunction and alteration of the gut microbiome as possible explanations for the link between PCOS and type 2 diabetes but these have not been extensively studied and validated when compared with the insulin resistance hypothesis [8].
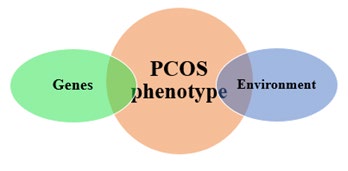
Familial clustering has been a recurrent decimal in various epidemiological studies looking at PCOS [9]. Genetic studies have identified certain candidate genes as the underlying factors responsible for the development of PCOS. These genes include genes involved in insulin signaling, genes involved in production of androgens and genes coding for inflammatory cytokines. Understanding the roles of these genes is believed to shed more light in the pathogenesis of PCOS. This may redirect the approach to diagnosing and managing PCOS. Although there is clustering of the diseases among families, however, phenotypic differences among the affected family members suggest that the disease is a result of interaction between the genotype and the environment and both are to be ‘blamed’. The gene-environment interaction is illustrated in Figure 1 below.
Epidemiology of PCOS
Irvin Freiler Stein and Michael Leventhal, through their seminal work in 1935, were the first to describe a clustering of amenorrhoea, hirsutism and polycystic ovaries in seven women [10]. Estimate of prevalence of PCOS is difficult to compare across studies due to the differences in the population studied, the year the study was done and the criteria adopted to define PCOS in each study. Prevalence of PCOS reported from various countries and the diagnostic criteria are illustrated in Table 1 below. Studies have shown that using the Rotterdam criteria gives twice the prevalence rate reported using the National Institute of Health (NIH) criteria.
| Country | Diagnostic criteria | Prevalence | |
|---|---|---|---|
| Ugwu, et al. 2013 [11] | Nigeria | Rotterdam criteria | 18.10% |
| Joshi, et al. 2015 [12] | India | Androgen Excess Society Criteria | 10.70% |
| March, et al. 2010 [13] | Australia | National Institute of Health criteria | 8.70% |
| Cahill, 2009 [14] | USA | Rotterdam criteria | 10% |
| Azziz, et al. 2011 [15] | UK | National Institute of Health criteria | 6-9% |
Table 1: Prevalence of PCOS across countries, using different criteria.
Risk factors
Family history of PCOS in first degree relatives is a risk factor for developing PCOS. Similarly, twin studies have reported a higher concordance rate among monozygotic twins compared with dizygotic twins. Other documented risk factors in the literature include background obesity, use of valproic acid (for seizure disorder). Interestingly, even though PCOS could predispose a woman to developing diabetes, background diabetes is a risk factor for PCOS [16]. So, the relationship between PCOS and diabetes is reciprocal. The reciprocal relationship is illustrated in Figure 2 below.
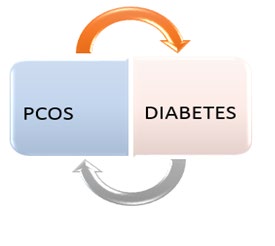
Insulin resistance alone cannot explain this relationship because PCOS is more common even in patients with type 1 diabetes, in whom insulin resistance is not a prominent aetiopathogenic factor. Extremes of birth weight have also been implicated as a risk factor for PCOS.
Diagnostic Criteria
There are four commonly quoted criteria for the diagnosis of PCOS and the components of each criteria scheme are summarized in Table 2 below. These criteria include the Rotterdam criteria, National institute of Health (NIH) criteria, European Society for Human Reproduction and Embryology (ESHRE) and the American Society for Reproductive Medicine (ASRM) criteria, Androgen excess and PCOS (AE-PCOS) society criteria and the Society of Obstetricians and Gynaecologists of Canada criteria.
The NIH criteria are produced from a conference of the National Institute of Child Health and Human Disease (NICHD) in 1990. It was the first widely accepted criteria scheme for the diagnosis of PCOS. In 2003, the European Society for Human Reproduction and Embryology and American Society for Reproductive Medicine formed a PCOS Consensus Workshop Group who proposed a set of criteria for the diagnosis of PCOS based on the agreements made in a conference held in Rotterdam. These criteria are commonly referred to as the Rotterdam criteria.
Rotterdam criteria are the most referenced criteria for the diagnosis of PCOS. The Rotterdam criteria is the most commonly referenced criteria and it is recommended by the Endocrine Society. In 2006, the Androgen excess and PCOS (AE-PCOS) society made a position statement on the diagnosis of PCOS and formalized their set of criteria in 2009. The Society of Obstetricians and Gynaecologists of Canada emphasized that conditions associated with androgen excess such as Cushing’s syndrome, congenital adrenal hyperplasia (CAH) and androgen-producing tumors must be excluded before a diagnosis of PCOS is made, just like the other criteria earlier mentioned. SOGC criteria are essentially similar to the Rotterdam criteria.
| Rotterdam | AE-PCOS | NIH | SOGC | |
|---|---|---|---|---|
| Criteria | □Clinical/Biochemical hyperandrogenism | □Clinical/Biochemical hyperandrogenism | □Clinical/Biochemical Hyperandrogenism | □Clinical/Biochemical hyperandrogenism |
| Criteria | □Menstrual dysfunction | □Menstrual Dysfunction and/or polycystic ovaries | □Menstrual dysfunction | □Menstrual dysfunction |
| Criteria | □Polycystic ovaries | □Polycystic ovaries | ||
| Exclusion | Other androgen excess disorders | Other androgen excess disorders | Other androgen excess disorders | Other androgen excess disorders |
| Recommendation | Any 2 of the criteria | The 2 criteria required | The 2 criteria required | Any 2 of the criteria |
Table 2: Diagnostic criteria for PCOS.
Pathophysiology
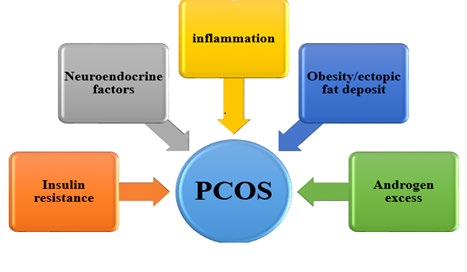
PCOS usually develops a few years after puberty. A major underlying factor is excess androgen secretion. Hyperinsulinaemia is believed to have contributed to excess androgens. Androgen excess causes the ovarian follicles to grow, creating the characteristic ultrasonographic finding of string-of-pearl morphology. In addition, there is an increased frequency of the pulsatile secretion of luteinizing hormone (LH) and increased LH/FSH ratio has been extensively described in the literature. This is thought to be due to increased gonadotrophin releasing hormone (GnRH) and this underlies the neuroendocrine aspect of PCOS pathophysiology.
PCOS is characterized by insulin resistance independent of obesity or androgen excess. Β-cells of the pancreas react to this insulin resistance by ensuring compensatory hyperinsulinaemia. Hyperinsulinaemia enhances ovarian androgen synthesis and lowers sex hormone binding globulin (SHBG). Lowered SHBG implies more free androgens available manifesting as hirsutism.
Hypersecretion of androgens by the stromal theca cells of the polycystic ovary leads to the cardinal clinical manifestation of the syndrome, hyperandrogenism, and is also one of the mechanisms whereby follicular growth is inhibited with the resultant excess of immature follicles. Moreover, in the presence of excess calories, fat cells undergo hypertrophy and/or hyperplasia. Insulin resistance ensures increased adipocytes lipolysis and trafficking of fatty acids to the liver leading to enhanced hepatic lipogenesis.
The developmental hypothesis states that the in- utero exposure of female foetal cells leads to cellular reprogramming which eventually causes PCOS [17]. This has been confirmed in animal studies. Also, alteration in the gastrointestinal tract microbiome is believed to also play a role in the development of PCOS. The pathophysiology of PCOS is illustrated in Figure 3 below.
The Roles of Genes in the Pathophysiology of PCOS
There is overwhelming evidence that genetic factors play a role in the pathophysiology of PCOS but the details of these roles are still subjects of intense research. Most of these genes are not transmitted in the Mendelian pattern but some studies on familial clustering suggest some Mendelian inheritance in some families [18]. Association studies and linkage studies are the most commonly employed methods of genetic studies on PCOS. In association studies, a particular allele or group of alleles are expected to be expressed more commonly in the affected population compared with the general population. Linkage is the tendency of genes to be transmitted as a cluster due to their proximity on the same chromosomes. Linkage studies are a genetic study technique that studies genes that are transmitted together in affected families in relations to the phenotypes. However, incomplete penetrance and variable expressivity have been the main drawbacks of genetic studies.
Studies on Familial Clustering
Several studies have demonstrated familial clustering in the genetic aetiopathogenesis of PCOS. However, lack of uniformity of expression between and within families has lent credence to the role of environmental factors. PCOS indeed aligns with the concept of nature versus nurture. Some researchers, working independently, have demonstrated an autosomal dominant patter in the family clusters in their studies although the penetrance was variable. The typical phenotypes in these studies were that of women manifesting with menstrual disturbance, clinical hyperandrogenism and polycystic ovaries. Interestingly, a male phenotype, manifesting with premature baldness and increased hairiness, have been reported. Other familial clustering studies could not demonstrate Mendelian inheritance but the phenotypes were similar.
Challenges of Genetic Studies
Lack of universally accepted diagnostic criteria makes it difficult to compare genotypes with phenotypes because various criteria use different phenotypes. Also, there are no clearly defined phenotypes for males and this makes general population studies difficult. Similarly, the sample size in such studies is usually small and inferences are difficult to be generalized. Randomization is difficult and linkage studies are difficult without clearly defined Mendelian pattern of inheritance. Moreover, incomplete penetrance and variable expressivity make it difficult to assign phenotype.
Genes Involved in Insulin Secretion
Hyperinsulinaemia is a common denominator in virtually all the phenotypes of PCOS hence, the various studies looking at genes involved in insulin secretion. Hyperinsulinaemia in PCOS has been reported to be due to a combination of insulin secretion and insulin action abnormalities. The insulin gene itself (INS) has been implicated in the pathophysiology of PCOS. The gene is located on the short arm of chromosome 11 [19]. Association studies have found an association between allelic variants of certain segments of insulin gene and PCOS. Calpain-10 is a protease that plays a dual role in the secretion and action of insulin. Variations in its genes have also been implicated in the pathophysiology of PCOS.
Genes Involved in Insulin Action
Insulin acts via the insulin receptor. The insulin receptor is a heterodimer and its gene is found on the short arm of chromosome 19. Several studies have reported an association between various mutations in the insulin receptor gene (INSR) and PCOS. A linkage study involving 367 families have also implicated the insulin receptor gene in the pathophysiology of PCOS [20]. Similarly, polymorphism in insulin receptor substrates genes (IRS 1 and IRS 2) has been associated with insulin resistance, a major factor in the development of PCOS [21].
Genes Involved in Androgens Biosynthesis
Excessive androgen synthesis causing hyperandrogenemia has been extensively reported in the literature. This dysregulation in androgen biosynthesis has some genetic basis. Conversion of cholesterol to pregnenolone is the first step in steroidogenesis. This reaction is catalyzed by an enzyme called side chain cleavage enzyme. Studies have shown an association between variations in the side chain cleavage enzyme gene and the development of PCOS [22]. Abnormalities in the genes of other enzymes involved in ovarian and adrenal steroidogenesis such as 17-α hydroxylase, 17,20-lyase and 21-hydoxylase have all been shown to play some roles in the aetiopathogenesis of PCOS. Aromatase is an enzyme that convers androgens to oestrogens thereby helping to regulate the concentration of androgens in the circulation. Its gene is found on the short arm of chromosome 15 [23]. Variations in certain segments of this gene have been linked with PCOS.
Genes Involved in Androgen Action
Androgens ultimately act via the androgen receptor. The androgen receptor gene (AR) is located on the short arm of chromosome X [24]. This is rich in CAG repeats and variations in these repeats have been associated with exaggerated response of the receptor. An abnormality of these repeats contribute to some PCOS phenotypes where the serum level of androgens is normal yet the patient is manifesting symptoms and signs of androgen excess [25].
Sex hormone binding globulin (SHBG) binds to the majority of circulating androgens and it determines the amount of free unbound androgens available to exert its physiological effects. SHBG is a homodimer and its gene is found in the short arm of chromosome 17 [26]. A functional polymorphism in the promoter region of the gene has been linked with reduced affinity of SHBG to androgen thereby increasing the amount of free androgens available to interact with the androgen receptor ultimately leading to hyperandrogenism [27]. This is thought to be the factor behind some phenotypes of PCOS.
Genes Involved in Gonadotrophins Physiology
The human gonadotropins are follicle stimulating
hormone (FSH) and luteinizing hormone (LH). They are both heterodimers with similar α subunit and different β subunits. They are produced in the anterior pituitary gland. FSH stimulates ovarian development while LH stimulates ovulation and ovarian androgen secretion. In PCOS, studies have shown an increased level of LH which is believed to be contributory to the anovulatory cycles observed in patients with PCOS. LH is a heterodimer (made up of α and β subunits) and the β subunit is responsible for the specificity of LH. A study has reported a higher prevalence of mutations of the LH β subunit gene in obese women with PCOS [28]. LH acts physiologically via the LH receptor. Activating mutation of LH receptor gene has been found in 5 families with several members manifesting the clinical features of PCOS. Activin is a glycoprotein produced by the ovarian follicles. Its physiologic effects include inhibition of FSH secretion in a negative feedback mechanism and inhibition of the action of LH on ovarian androgen secretion. Follistatin is a high-affinity binding protein for activin. Overexpression of follistatin gene leads to reduced activity of activin. Reduced activity of activin implies that FSH stimulation of follicles is unregulated thereby causing arrested ovarian folliculogenesis which manifests as polycystic ovaries [29]. Similarly, the androgen secretion-enhancing effect of LH is increased thereby contributing to hyperandronism. Studies have reported overexpression of follistatin gene in certain phenotypes of PCOS [30].
Genes Involved in Adipose Tissue Physiology
Peroxisome proliferator-activated receptor γ (PPAR-γ) is a transcription factor whose physiological role is mainly lipogenesis. Polymorphisms of PPAR-γ are associated with the development of PCOS. Thiazolidinediones, which activate t PPAR-γ, has been used successfully in the management of PCOS which also lends credence to the fact that PPAR-γ gene has a role to play in the disease. Regarding PCOS, studies on the polymorphisms of leptin and adiponectin genes have yielded inconsistent findings. Genes Involved in Chronic Inflammation Chronic subclinical inflammation plays a role in the pathophysiology of PCOS. Studies on the association between PCOS and polymorphisms of tumor necrosis factor-α (TNF-α) and its receptor as well as interleukin 6 (IL-6) and its receptor too have yielded inconsistent findings. While some researchers have reported a significant association, others have not. Genes involved in the aetiopathogenesis of PCOS are summarized in Table 3 below.
| Examples of genes | |
|---|---|
| Genes involved in insulin secretion | INS |
| Genes involved in insulin action | INSR, IRS-1, IRS-2 |
| Genes involved in androgen biosynthesis | CYP 111α, CYP 21, CYP 17 |
| Genes involved in androgen action | AR, SHBG |
| Genes involved in gonadotropins physiology | LHβ, follistatin |
| Genes involved in adipose tissue physiology | PPAR-γ |
| Genes involved in chronic inflammation | TNF-α, IL-6 |
Table 3: Genes involved in the pathophysiology of PCOS.
Conclusion
Polycystic ovarian syndrome is a common disorder among women of reproductive age group. The pathophysiology is still a subject of intense research. It arises from to the interactions between the environment and the predisposing genotypes. Genes mediating insulin resistance, androgen and gonadotropin action and chronic inflammation have all been implicated in the pathophysiology of PCOS. While scientific efforts are being intensified to further characterize the genetic basis of the disease, it must be emphasized that genetic studies have a lot of challenges.
References
-
Witchel SF, Oberfield SE, Pena AS (2019) Polycystic Ovary Syndrome: Pathophysiology, Presentation, and Treatment with Emphasis on Adolescent Girls. JES 3(8): 1545-1573.
-
Escobar-Morreale HF (2008) Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol 14(5): 270-284.
-
Palomba S, de Wilde MA, Falbo A, Koster MPH, La Sala GB, et al. (2015) Pregnancy complications in women with polycystic ovary syndrome: new clinical and pathophysiological insights. Hum Reprod Update 21(5): 575-592.
-
Esan A, Azeez TA (2020) Challenges of Glycemic Control in COVID-19 Patients with Diabetes Mellitus in Resource- poor Settings. IJIRMS 5(8): 329-333.
-
Palomba S, Santagni S, Falbo A, Battista La Sala G (2015) Complications and challenges associated with polycystic ovary syndrome: current perspectives. Int J Womens’ Health 7: 745-763.
-
Wang ET, Calderon-Margalit R, Cedars MI (2011) Polycystic ovary syndrome and risk for long-term diabetes and dyslipidemia. Obstet Gynecol 117(1): 6-13.
-
Azeez TA (2020) Hypomagnesemia and insulin resistance: gaining better understanding of the pathophysiology of type 2 diabetes. Insights Biomed 5(4): 22.
-
Pani A, Gironi I, Di Vieste G, Mion E, Bertuzzi F, et al. (2020) From Prediabetes to Type 2 Diabetes Mellitus in Women with Polycystic Ovary Syndrome: Lifestyle and Pharmacological Management. Int J Endocrinol 2020: 6276187.
-
Deligeoroglou E, Kouskouti C, Christopoulus P (2009) The role of genes in the polycystic ovary syndrome: Predisposition and mechanisms. Gynaecol Endocrinol 25(9): 603-609.
-
Sirmans SM, Pate KA (2014) Epidemiology, diagnosis, and management of polycystic ovary syndrome. Clin Epidemiol 6: 1-13.
-
Ugwu GO, Iyoke CA, Onah HE, Mba SG (2013) Prevalence, presentation and management of polycystic ovary syndrome in Enugu, south east Nigeria. Niger J Med 22(4): 313-316.
-
Joshi B, Mukherjee S, Patil A, Purandare A, Chauhan S, et al. (2014) A cross-sectional study of polycystic ovarian syndrome among adolescent and young girls in Mumbai, India. Indian J Endocrinol Metab 18(3): 317-324.
-
March WA, Moore VM, Willson KJ, Phillips DI, Norman RJ, et al. (2010) The prevalence of polycystic ovary syndrome in a community sample assessed under contrasting diagnostic criteria. Hum Reprod 25(2): 544- 551.
-
Cahill DJ (2009) PCOS. BMJ Clin Evid 2009: 1408.
-
Azziz R, Dumesic DA, Goodarzi MO (2011) Polycystic ovary syndrome: An ancient disorder?. Fertil Steril 95(5): 1544-1548.
-
Escobar-Morreale HF, Roldán B, Barrio R, Alonso M, Sancho J, et al. (2000) High prevalence of the polycystic ovary syndrome and hirsutism in women with type 1 diabetes mellitus. J Clin Endocrinol Metab 85(11): 4182- 4187.
-
Abbott DH, Dumesic DA, Franks S (2002) Developmental origin of polycystic ovary syndrome-a hypothesis. J Endocrinol 174(1): 1-5.
-
Unlutyrk U, Harmanci A, Kocaefe C, Yildiz BO (2007) The Genetic Basis of the Polycystic Ovary Syndrome: A Literature Review Including Discussion of PPAR-γ. PPAR-γ Res 2007: 49109.
-
Junien C, van Heyningen V (1990) Report of the committee on the genetic constitution of chromosome 11. Cytogenet Cell Genet 55(1-4): 153-169.
-
Urbanek M, Woodroffe A, Ewens KG, Diamanti- Kandarakis E, Legro RS, et al. (2005) Candidate gene region for polycystic ovary syndrome on chromosome 19p13.2. J Clin Endocrinol Metab 90(12): 6623-6629.
-
Jellema A, Zeegers MP, Feskens EJ, Dagnelie PC, Mensink RP (2003) Gly972Arg variant in the insulin receptor substrate-1 gene and association with Type 2 diabetes: a meta-analysis of 27 studies. Diabetologia 46(7): 990- 995.
-
Diamanti-Kandarakis E, Bartzis MI, Bergiele AT, Tsianateli TC, Kouli CR (2000) Microsatellite polymorphism (tttta) (n) at -528 base pairs of gene CYP11alpha influences hyperandrogenemia in patients with polycystic ovary syndrome. Fertil Steril 73(4): 735-741.
-
Chen SA, Besman MJ, Sparkes RS, Zollman S, Klisak I, et al. (1988) Human aromatase: cDNA cloning, Southern blot analysis, and assignment of the gene to chromosome 15. DNA 7(1): 27-38.
-
Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, et al. (1988) Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science 240(4850): 327-330.
-
Mifsud A, Ramirez S, Yong EL (2000) Androgen receptor gene CAG trinucleotide repeats in anovulatory infertility and polycystic ovaries. J Clin Endocrinol Metab 85(9): 3484-3488.
-
Selby C (1990) Sex hormone binding globulin: origin, function and clinical significance. Ann Clin Biochem 2 (Pt 6): 532-541.
-
Xita N, Tsatsoulis A, Chatzikyriakidou A, Georgiou I (2003) Association of the (TAAAA)n repeat polymorphism in the sex hormone-binding globulin (SHBG) gene with polycystic ovary syndrome and relation to SHBG serum levels. J Clin Endocrinol Metab 88(12): 5976-5980.
-
Rajkhowa M, Talbot JA, Jones PW, Pettersson K, Haavisto AM, et al. (1995) Prevalence of an immunological LH beta-subunit variant in a UK population of healthy women and women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 43(3): 297-303.
-
Franks S, Gharani N, McCarthy M (2001) Candidate genes in polycystic ovary syndrome. Hum Reprod Update 7(4): 405-410.
-
Legro RS, Spielman R, Urbanek M, Driscoll D, Strauss JF 3rd, et al. (1998) Phenotype and genotype in polycystic ovary syndrome. Recent Prog Horm Res 53(4): 217-256.
- Shaping Healthy Futures: Pediatric Endocrine Breakthroughs of 2025
- Precision Medicine in Obesity: Customizing Treatment for 2025
- The Thyroid Revolution: How 2025 is Redefining Hormone Health
- Editorial- Targeting Immunometabolism for Generating Innovative Therapies for Cancer
- Current Knowledge of Chickenpox
- Correlation of Preinjection Values of Gonadotropins and Estradiol Level with Clinical and Radiologic Evidence of Sufficient Pubertal Suppression in Girls with Central Precocious Puberty