Cloves Syndrome: a Rare Overgrowth Disorder
Overgrowth syndromes are characterized by Global or localized excessive growth coupled with additional abnormalities, such as vascular malformations and neurological and/or visceral disorders [1]. Cloves syndrome is a recently described overgrowth syndrome caused by a mutation of an allele of the PIK3CA gene [2,3]. It is an acronym for congenital lipomatous overgrowth, vascular malformations, epidermal nevi and skeletal anomalies, scoliosis and spinal anomalies [3]. It is characterized by an asymmetrical body hypertrophy with macrodactyly and scoliosis, a distinctive thoraco-abdominal congenital lipomatous hamartoma, and various malformations, particularly vascular malformations including capillary, venous, lymphatic micro or macrocystic malformations, and rarely arteriovenous malformations, often located opposite the lipomatous masses [1,2]. The syndrome also presents with an epidermal nevus following a linear path along Blaschko’s lines [1,2], as well as neurological anomalies such as hemimegalencephaly and corpus callosum dysgenesis, which are associated with varying degrees of intellectual impairment [3,4], and visceral malformations like renal agenesis/hypoplasia and splenic lesions [1,2]. Primary differential diagnosis is Proteus syndrome, which has a higher morbidity and mortality rate compared to Cloves syndrome. Proteus syndrome is characterized, unlike Cloves syndrome, by acquired lipomatous masses and a cerebriform aspect of the soles in 71% of cases, and it often manifests between the ages of 6 and 18 months [2,4]. The diagnosis of Cloves syndrome is based on clinical presentation [3] and confirmed through genetic testing [1]. Treatment is primarily symptomatic and may include surgery to address overgrowth and physical therapy to improve mobility [3]. Inhibitors of mTOR (mammalian target of rapamycin) and PI3K (phosphoinositide 3-kinase) have shown promising results as treatment options for Cloves syndrome. However, clinical trials and studies evaluating their efficacy are currently limited.
El-Ammari S*, Baybay H, Elloudi S, Soughi M, Douhi Z and Mernissi FZ
Dear Editor,
Overgrowth syndromes are characterized by Global or localized excessive growth coupled with additional abnormalities, such as vascular malformations and neurological and/or visceral disorders [1]. Cloves syndrome is a recently described overgrowth syndrome caused by a mutation of an allele of the PIK3CA gene [2, 3]. It is an acronym for congenital lipomatous overgrowth, vascular malformations, epidermal nevi and skeletal anomalies, scoliosis and spinal anomalies [3]. It is characterized by an asymmetrical body hypertrophy with macrodactyly and scoliosis, a distinctive thoraco-abdominal congenital lipomatous hamartoma, and various malformations, particularly vascular malformations including capillary, venous, lymphatic micro or macrocystic malformations, and rarely arteriovenous malformations, often located opposite the lipomatous masses [1, 2]. The syndrome also presents with an epidermal nevus following a linear path along Blaschko’s lines [1, 2], as well as neurological anomalies such as hemimegalencephaly and corpus callosum dysgenesis, which are associated with varying degrees of intellectual impairment [3, 4], and visceral malformations like renal agenesis/hypoplasia and splenic lesions [1, 2]. Primary differential diagnosis is Proteus syndrome, which has a higher morbidity and mortality rate compared to Cloves syndrome. Proteus syndrome is characterized, unlike Cloves syndrome, by acquired lipomatous masses and a cerebriform aspect of the soles in 71% of cases, and it often manifests between the ages of 6 and 18 months [2, 4]. The diagnosis of Cloves syndrome is based on clinical presentation [3] and confirmed through genetic testing [1]. Treatment is primarily symptomatic and may include surgery to address overgrowth and physical therapy to improve mobility [3]. Inhibitors of mTOR (mammalian target of rapamycin) and PI3K (phosphoinositide 3-kinase) have shown promising results as treatment options for Cloves syndrome. However, Letter to Editor clinical trials and studies evaluating their efficacy are currently limited [4, 5].
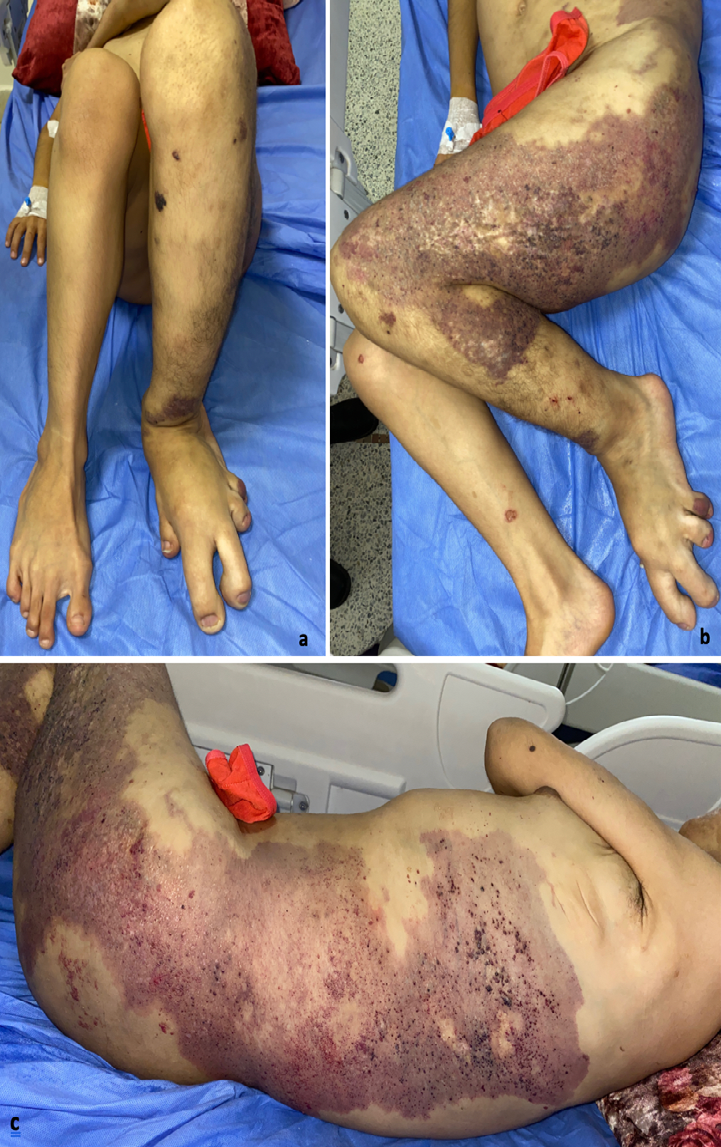
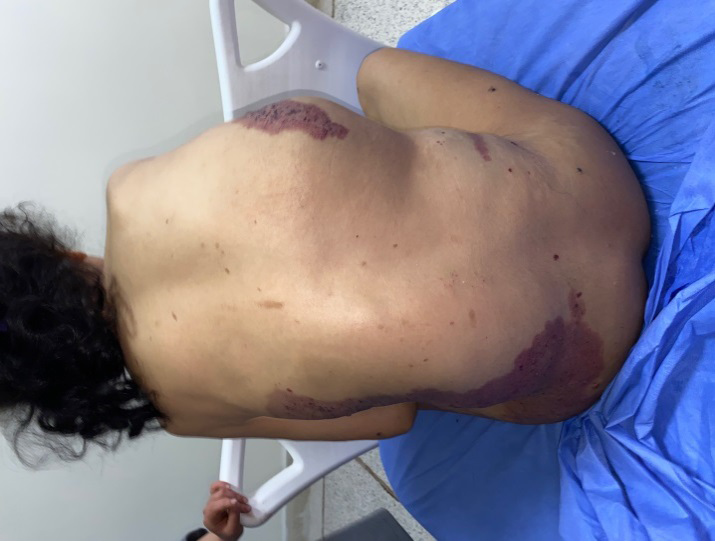
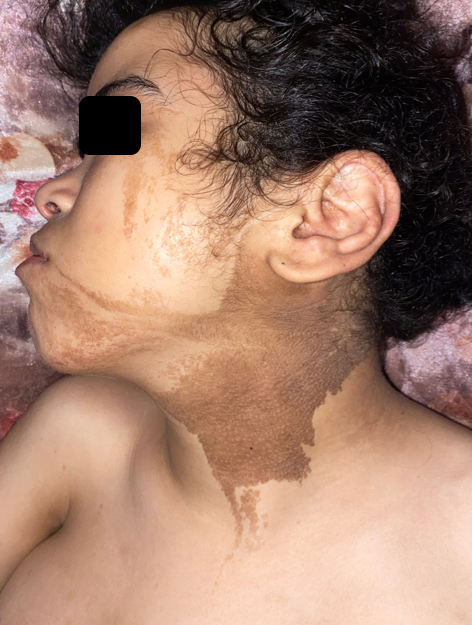
A 19-year-old girl from a non-consanguineous marriage with a history of left ear deafness and recurrent otitis presented with angiomatous spots on the left side of her body since birth. These spots progressively increased in size, along with the growth of a dorsal mass and the volume and length of her left lower limb. Over time, small liquid lesions with either hemorrhagic or clear contents appeared on the angiomatous lesions. Examination revealed left facial and lingual hemihypertrophy, as well as hypertrophy of the left lower limb, resulting in inequality between the two lower limbs with macrodactyly of the second and third toes of the left foot, along with foot deformity (Figure 1a). The patient exhibited also a macular angiomatous plaque extending from the upper third of the left lateral surface of the trunk to the upper third of the left leg, with multiple vesicles containing hemorrhagic or clear fluid, indicating lymphangiectasias (Figures 1b, c). Similar angiomatous plaques were observed on the anterior surface of the left leg, the toes on the same side, and the upper third of the right lateral aspect of the thorax. A well-defined soft mass measuring 15 cm in diameter was also present (Figure 2). Additionally, a linear verrucous epidermal nevus was observed, extending from the left half of the nape of the neck to the left cheek and chin (Figure 3). A spinal X-ray confirmed the presence of scoliosis, while an ultrasound (USG) confirmed the lipomatous origin of the mass on the right lateral chest wall. The echodoppler examination of the limbs did not reveal any arteriovenous malformations, and a cerebral MRI showed a malformative triventricular hydrocephalus associated with aqueductal stenosis. Genetic testing was not conducted due to limited resources. The patient had previously been diagnosed with intracranial hypertension, specifically neuromeningeal tuberculosis, and had started treatment with anticapillaries but was subsequently lost to follow-up.
References
-
Martinez-Lopez A, Blasco-Morente G, Perez-Lopez I, Herrera-Garcia JD, Luque-Valenzuela M, et al. (2017) CLOVES syndrome: review of a PIK3CA-related overgrowth spectrum (PROS). Clin Genet 91(1): 14-21.
-
Guillet A, Aubert H, Tessier MH, David A, Perret C, et al. (2014) Syndrome CLOVES : un syndrome malformatif proche du syndrome de Protée. Ann Dermatol Venereol 141(8-9): 507-513.
-
Mahajan VK, Gupta M, Chauhan P, Mehta KS (2019) Cloves Syndrome: A Rare Disorder of Overgrowth with Unusual Features - An Uncommon Phenotype? Indian Dermatol Online J 10(4): 447-452.
-
Manor J, Lalani SR (2020) Overgrowth Syndromes- Evaluation, Diagnosis, and Management. Front Pediatr 8: 574857.
-
Alexisa L, Yasera D, Laura TL (2022) Efficacy and Safety of Long-term Sirolimus Use as Part of Multidisciplinary Care in a Pediatric Patient with CLOVES Syndrome: Case Report. Translational Science of Rare Diseases 6(1-2): 25-32.
- Epithelioid Granuloma; 3cases with Different Clinical Features
- Advancing Representation in Dermatology Clinical Trials: Ethical, Scientific, and Regulatory Imperatives for Inclusion Across all Fitzpatrick Skin Types
- A Case of Atopic Dermatitis with Concurrent Psoriasis Vulgaris: Successful Treatment with Upadacitinib
- Innovation Lifting Eyeshadow: A Synthesis of Makeup and Optical Illusion
- Distinguishing Superficial Actinic Porokeratosis from Actinic Keratosis with UVF Dermoscopy: A Case Report
- High Mobility Group Box 1 (HMGB1) in Cutaneous Inflammation: An Immune Modulator Bridging Cellular Stress, Ferroptosis and Danger Signaling