Epidermolysis Bullosa Acquisita Presenting a Mix of Classical and Atopiform Features
Epidermolysis bullosa acquisita (EBA) is a rare acquired autoimmune bullous disorder. This disease is characterised by the presence of autoantibodies targeted against Collagen VII of the anchoring fibrils in the basement membrane zone of skin and mucous membranes. The two most common presentations of EBA are the classical or mechano-bullous and the bullous pemphigoid (BP) like forms. Patients with BP-like form of EBA have generally profuse skin lesions suggestive of BP in some areas and EBA in others. We present a case of inflammatory BP like EBA, in a young woman, who presented with classical and also atypical atopiform features.
Introduction
Epidermolysis bullosa acquisita (EBA) is an acquired, rare, autoimmune subepidermal blistering disease involving skin and mucous membranes, and generally occurs in adulthood. It is characterized by autoantibodies targeting collagen VII of the anchoring fibrils in the basement membrane zone of skin and mucous membrane. The immune-mediated disruption of the anchoring fibrils likely contributes to cleavage within the basement membrane zone and clinical blistering. The classic mechanobullous type of EBA is characterized by skin fragility, noninflammatory tense bullae, milia, and scarring. The inflammatory form of EBA presents with generalized vesiculobullous eruption primarily on the trunk and flexural areas, mimicking bullous pemphigoid. It is a rare disorder with an estimated incidence reported to be less than 0.5 per million [1, 2]. Two onset age peaks are reported; the second and seventh decades [3].
This case is presented because of its rarity, interesting aggravating factors, unusual presentation, in the form of palmoplantar involvement and generalized lichenification and a rapid and complete response to dapsone, despite its severity, and the utility of low dose oral corticosteroids as a rescue medicine.
A 17-year-old female presented with a history of recurrent generalised blistering since birth. None of the family members, including parents, six sisters and a brother were affected. There was no personal or family history suggestive of atopy and the IgE was within normal limits. There were no symptoms related to bowel habits. The blisters were associated with severe itching and exacerbations were noted during summer. There was no preceding history of trauma, other than scratching in the majority of lesions. Patient gave a history of flushing of the face and generalized discomfort in warm conditions.
Dermatological examination revealed multiple generalised bullae over the chest, back, neck, forearm, legs and the palms and soles (Figure 1). Some of the bullae were linear and some showed angulated outlines (Figure 2). There were crusted erosions and mild scarring in several areas. Palms and soles showed collapsed bullae, and thickened, hyperkeratotic skin (Figures 3 & 4).
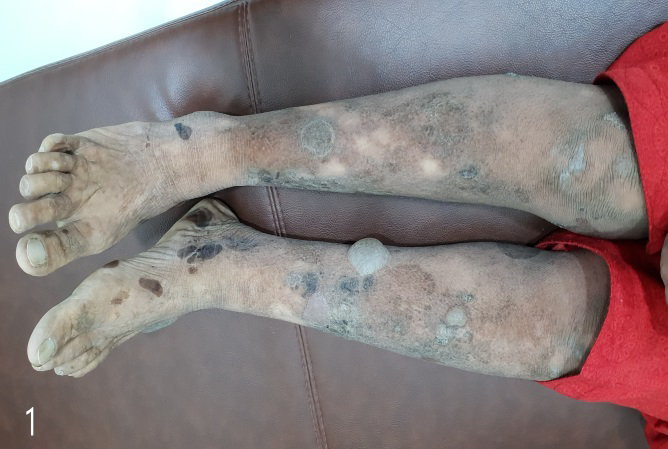
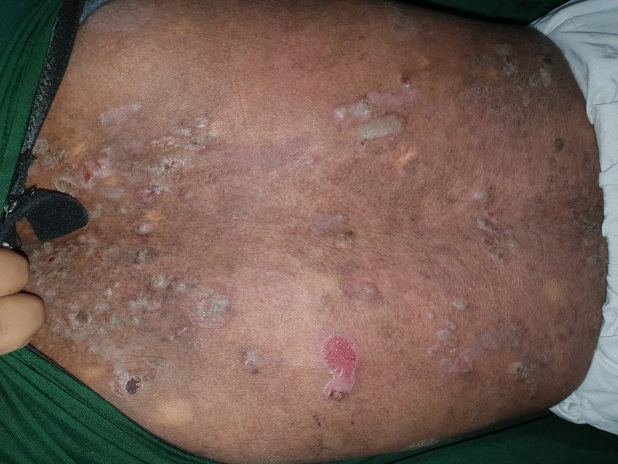


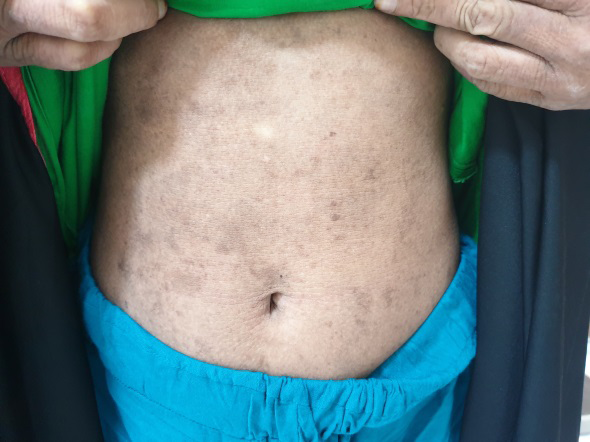
Entire skin showed xerosis and lichenification (Figure 5). The mucosae, hair and nails were normal. Serum ELISA test for BP 180 and BP 230 was negative, ruling out bullous pemphigoid. Provisional diagnosis of linear IgA disease and epidermolysis bullosa simplex was made, considering the clinical picture.
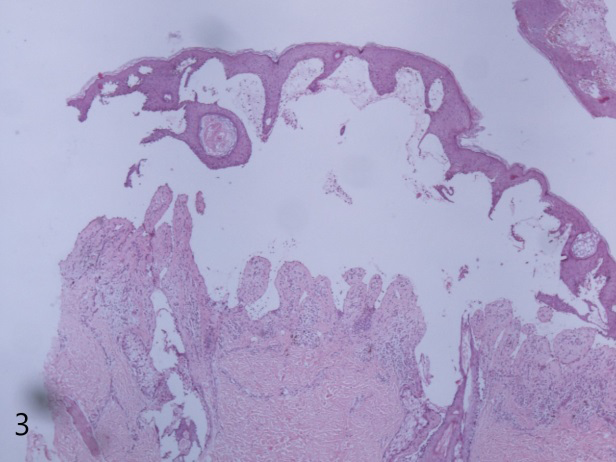
Histopathological examination revealed a subepidermal bulla, and a pseudoepitheliomatous hyperplasia with irregular downward projections of the epithelium. Dermis showed mild to moderate perivascular lymphocytes with occasional neutrophils and eosinophils (Figure 6).
Direct immunofluorescence study showed linear deposits of IgG and C3 along the basement membrane zone. IgA and IgM were negative (Figure 7). Salt split preparation showed deposition of IgG along the floor of the subepidermal bulla (Figure 8).
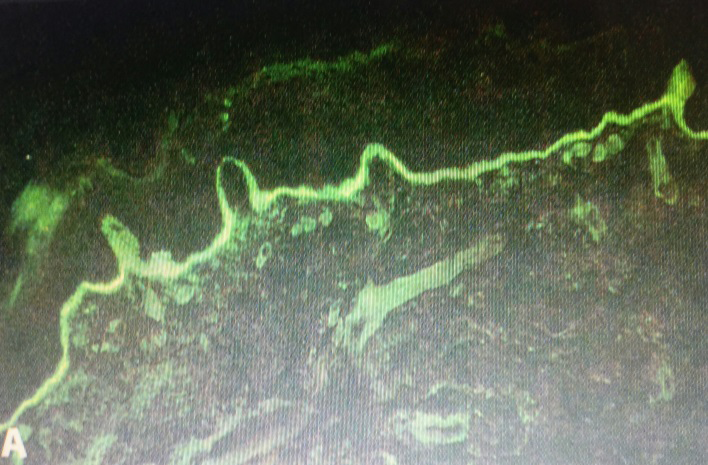
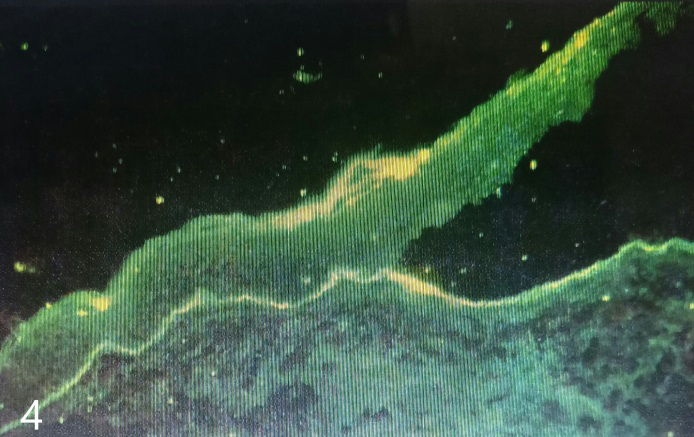
The clinical picture, the histopathology showing a subepidermal bulla, and the direct immunofluorescence study, including the salt split preparation, led us to the diagnosis of EBA. Patient was put on Tablet Dapsone 100 mg daily after the requisite blood work. The patient started responding from 4th week and there was a complete remission at week 12. After 2 months, fresh bullae began appearing, in spite of continuing dapsone. Low dose prednisolone 10 mg daily was started, in addition to dapsone. Disease remitted in 6 weeks, and steroids were gradually withdrawn over the next 6 weeks. The patient is still maintained on dapsone 100 mg daily, and is in complete remission since the last 4 months.
Discussion
EBA is a very rare disorder. Clinically, it may resemble dystrophic epidermolysis bullosa, bullous pemphigoid, cicatricial pemphigoid or linear IgA dermatitis. Diagnosis rests upon the investigations. In our case histopathology showed a subepidermal bulla. DIF revealed a linear deposition of IgG along the basement membrane zone, ruling out linear IgA disease. Since our patient was only 17 years old, bullous pemphigoid was unlikely, and hence salt split preparation was done demonstrating the location of IgG to be the floor of the bulla.
International Bullous Diseases Group, IBDG updated consensus criteria for EBA diagnosis were published in 2017. They laid down a combination of criteria for a definitive diagnosis of EBA [4]. They concluded that criteria 1,2,3 and 9 would lead to a definitive diagnosis of EBA, viz., (1) a bullous disorder within the defined clinical spectrum; (2) histopathology revealing a subepidermal or subepithelial blister; (3) a positive DIF microscopy of perilesional skin or MM with linear IgG, C3, IgA and/or IgM deposits within the epithelial BMZ and (9) dermal labelling by DIF and/or IIF on SSS. Our case fulfilled the criteria laid down by the IBDG. Moreover, Gammon et al have reported 100% accuracy in the diagnosis of EBA by indirect immunofluorescence microscopy (IIF) using 1M NaCl split skin (SSS) as a substrate [5].
The patient presented with a clinical picture very similar to bullous pemphigoid. Analogous to bullous and mucous membrane pemphigoid, an EBA is considered severe if the patient has 10 or more cutaneous bullous lesions [6].
The patient presented with about 15 to 16 bullae, besides crusted erosions and collapsed bullae in the first visit. Our patient also showed significant palmoplantar involvement with bullae and marked hyperkeratosis. In addition, there was generalized xerosis and lichenification, especially affecting the flexures. Warm weather and increased humidity caused the disease to aggravate, suggesting another trigger in the pathogenesis of this disease. EBA patients respond with difficulty and that too partially. High doses of steroids and a combination of treatments have been tried earlier with variable success. Dapsone has been considered to be a safe and relatively effective first line treatment [7, 8, 9].
In the present patient, though the disease was severe, response to dapsone was prompt and complete initially. Later a small dose of corticosteroids, as a rescue medicine, controlled the relapse. The patient is now being maintained in complete remission by only dapsone 100 mg per day. We are reporting this case because of its rarity, interesting aggravating factors and unusual presentation, in the form of palmoplantar involvement and generalized lichenification and the gratifying response to dapsone, despite its severity, and the utility of low dose oral corticosteroids as a rescue medicine.
References
-
Bertram F, Brocker EB, Zillikens D, Schmidt E (2009) Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges 7(5): 434-440.
-
Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA (2004) Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol 43(12): 876-881.
-
Hubner F, Recke A, Zillikens D, Linder R, Schmidt E (2016) Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol 136(12): 2495-2498.
-
Squarcioni CP, Caux F, Schmidt E, Jonkman MF, Vassileva S, et al. (2017) International Bullous Diseases Group: consensus on diagnostic criteria for epidermolysis bullosa acquisita. Br J Dermatol 179(1): 30-41.
-
Gammon WR, Kowalewski C, Chorzelski TP, Kumar V, Briggaman RA, et al. (1990) Direct immunofluorescence studies of sodium chlorideseparated skin in the differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. J Am Acad Dermatol 22 (4): 664-670.
-
Koga H, Squarcioni CP, Iwata H, Jonkman MF, Ludwig RJ, et al. (2019) Epidermolysis Bullosa Acquisita: The 2019 Update. Front. Med 5: 362.
-
Kim JH, Kim YH, Kim SC (2011) Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol 91(3): 307-312.
-
Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, et al. (2010) Epidermolysis bullosa acquisita: what’s new? J Dermatol 37(3): 220-30.
-
Hashimoto T, Ishii N, Ohata C, Furumura M (2012) Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol 228 (1): 1-7.
- Epithelioid Granuloma; 3cases with Different Clinical Features
- Advancing Representation in Dermatology Clinical Trials: Ethical, Scientific, and Regulatory Imperatives for Inclusion Across all Fitzpatrick Skin Types
- A Case of Atopic Dermatitis with Concurrent Psoriasis Vulgaris: Successful Treatment with Upadacitinib
- Innovation Lifting Eyeshadow: A Synthesis of Makeup and Optical Illusion
- Distinguishing Superficial Actinic Porokeratosis from Actinic Keratosis with UVF Dermoscopy: A Case Report
- High Mobility Group Box 1 (HMGB1) in Cutaneous Inflammation: An Immune Modulator Bridging Cellular Stress, Ferroptosis and Danger Signaling