Chanarin-Dorfman Syndrome: The Enormous Value of a Simple Peripheral Blood Smears in Ichthyosis Saving Time and Money
Chanarin-Dorfman syndrome (CDS) is a rare non-lysosomal autosomal recessive neutrophil lipid storage disorder characterized by deposition of cytoplasmic neutral lipid droplets in many cell types. It is clinically characterized by nonbullous congenital ichthyosiform erythroderma, hepatomegaly, myopathy, ataxia, neurosensory hearing loss and varying developmental delay. A peripheral blood smear showing lipid droplets in granulocytesis highly suggestive of the diagnosis.
Ganjoo Shikhar1 and Ganjoo Tulika2
diagnosis.
Case Report
A 14 month old, developmentally normal for age male child, born out of second degree consanguineous marriage presented to us with complaints of upper abdominal distension for three weeks, dry skin with peeling since neonatal period and strabismus. On examination the child had generalized ichthyosis and hepatomegaly (Figure 1). There was no ataxia, nystagmus, or deafness or clinical signs of peripheral neuropathy or myopathy. Similar skin lesions were present in his cousin. We considered the differentials of non-bullous congenital icthyosiform erythroderma, lamellar ichthyosis and Chanarin-Dorfman syndrome.
On investigation peripheral blood smear revealed presence of vacuolated neutrophils (Jordan’s anomaly). (Figure 2) Liver enzymes were mildly elevated (alanine transaminase 93 IU/L and aspartate transaminase 112 IU/L) with normal bilirubin levels. Lipid profile was normal. Skin biopsy was consistent with non bullous icthyosiform erythroderma (Figure 3) with presence of vacuolated keratinocytes and vacuolated cells in eccrine gland (Figure 4). Liver biopsy showed macrovesicular steatosis with features of cirrhosis and steatohepatitis (Figure 5). The skeletal and ophthalmological evaluation was normal.
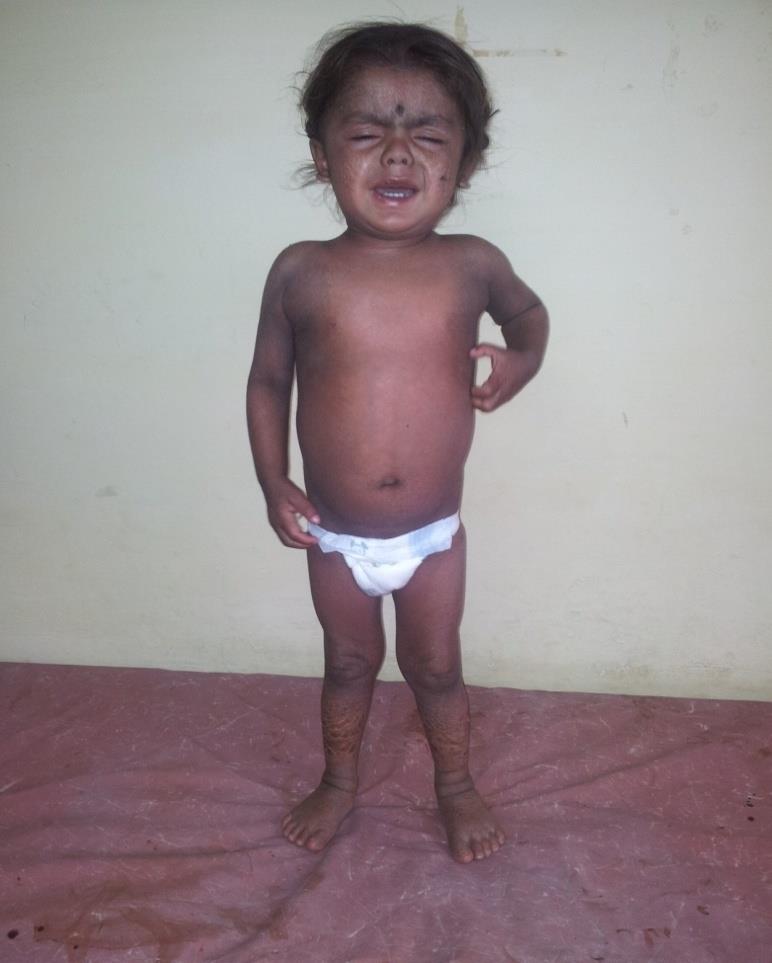
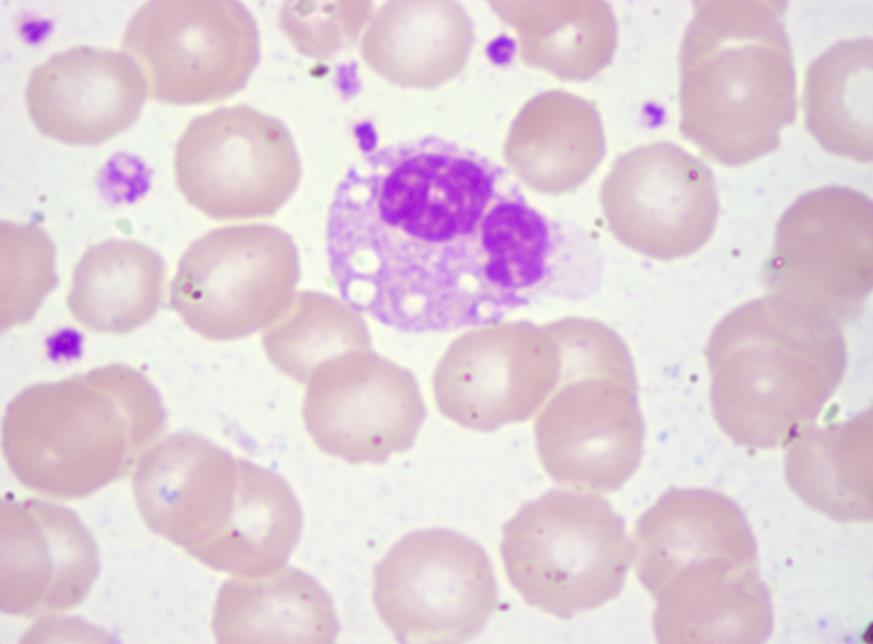
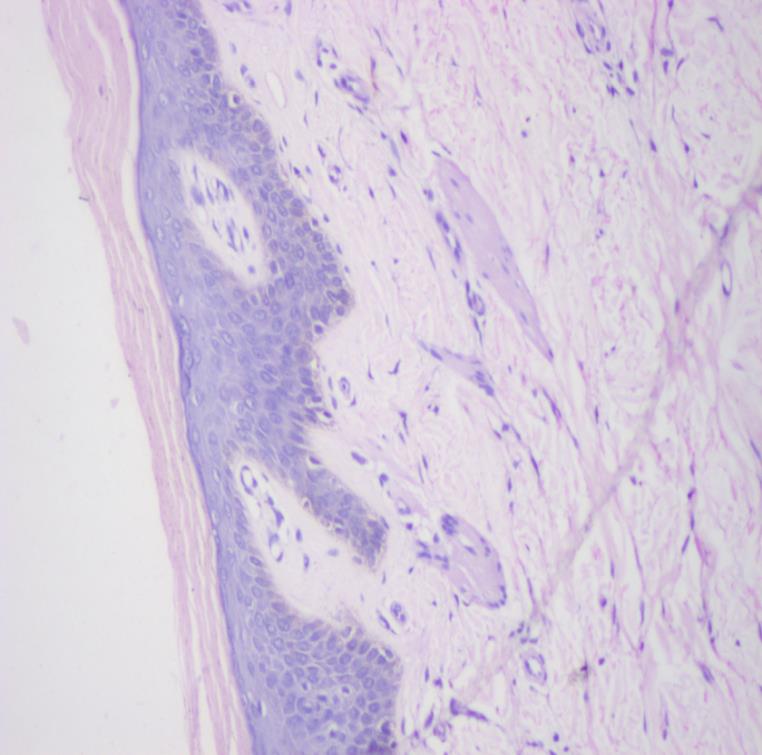
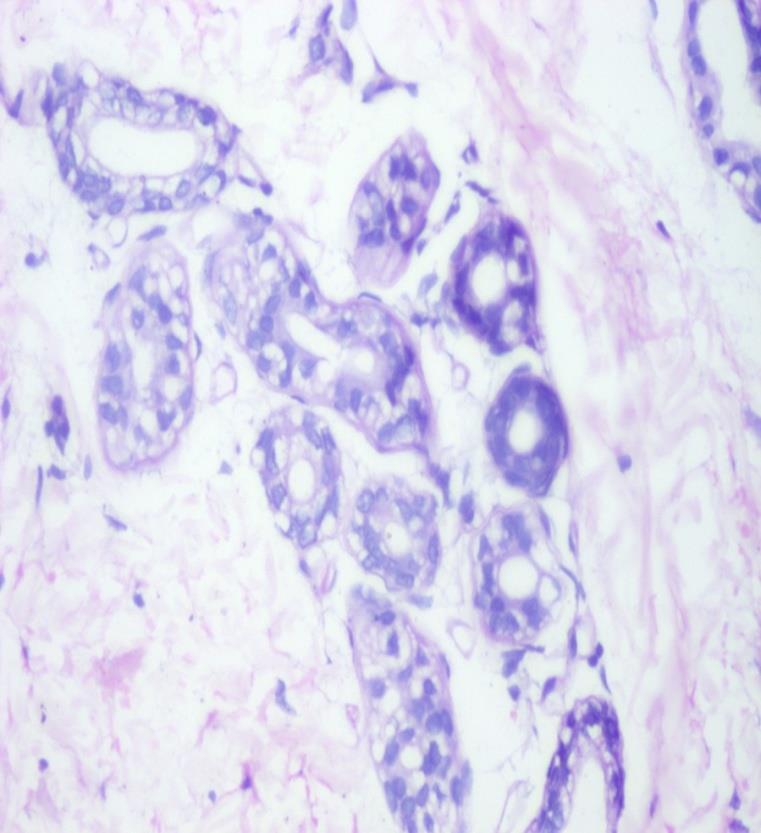
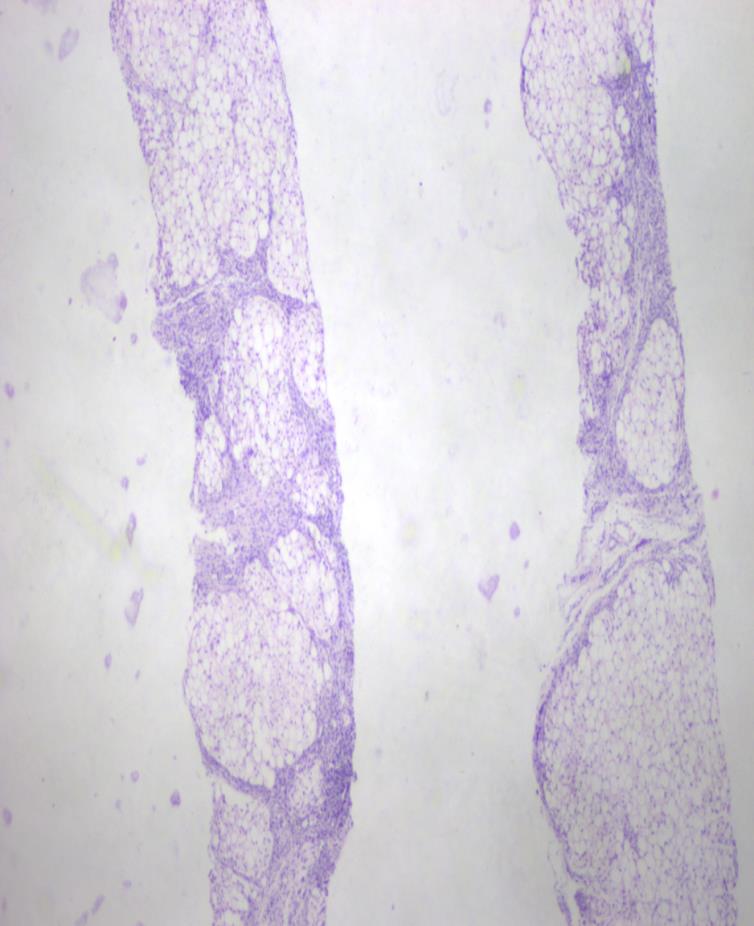
Figure 5: HPE of liver showing macrovescicular steatosis. (H & E 10X). With these typical featuresa diagnosis of CDS was considered. Isotretinoin 10 mg twice a week and emollients were prescribed along with a diet rich in medium chain triglycerides and deficient in long chain fatty acids. Follow up of the patient at 4 and 8 weeks showed progressively significant improvement in cutaneous lesions and normalization of liver function tests. Thus, a simple peripheral blood smear should be done to rule out CDS in any child presenting with ichthyosis and systemic involvement.
Discussion
Chanarin-Dorfman syndrome is a rare autosomal recessive metabolic disorder with deposition of cytoplasmic neutral lipid droplets in many cell types, in particular hepatocytes, keratinocytes and granulocytes. CDS is characterized by nonbullous congenital ichthyosiform erythroderma, hepatomegaly and varying degree of developmental delay. Muscle weakness (or myopathy), ataxia, neurosensory hearing loss, subcapsular cataracts and nystagmus can be present [1].
Expression of the syndrome is variable, but the diagnosis can be easily confirmed by a peripheral blood smear, which shows lipid droplets in granulocytes, known as Jordan’s anomaly [2]. A mutation in the ABHD5 gene on chromosome 3p21 is the genetic defect responsible for CDS [3]. Mutation of ABHD5 leads to deposition of triglycerides as lipid droplets in various organs due to loss of function of adipose triglyceride lipase enzyme [4, 5]. Our patient presented classically with congenital nonbullous icthyosiform erythroderma, hepatomegaly, strabismus and peripheral blood smear showed Jordan’s anomaly. Histopathological examination revealed deposition of lipid vacuoles in hepatocytes, keratinocytes, and eccrine glands. Thus the presence of lipid droplets in granulocytes in the setting of typical cutaneous features of generalized ichthyosis and hepatomegaly since the neonatal period is highly suggestive of Chanarin-Dorfman syndrome. We went ahead and did a skin biopsy to document the presence of similar changes in keratinocytes and eccrine glands and furthermore a liver biopsy that also confirmed macrovesicular steatosis with features of cirrhosis and steatohepatitis. This was done to confirm our speculation that it indeed was a patient of Chanarin-Dorfman syndrome but doing the same took us a lot of time and extra monetary spending was also involved. It did not contribute much in terms of diagnosis which we had highly suspected initially nor changed anything in terms of the management that we offered the patient. Thus we highly recommend that in a resource poor setting or where there is a need to establish diagnosis quickly in patients, the above set of clinical features with the presence of Jordan’s anomaly can be taken as a confirmatory test. This will go a long way in saving time and money of the patient.
References
-
Srebrnik A, Brenner S, Ilie B, Messer G (1998) Dorfman–Chanarin syndrome: morphologic studies and presentation of new cases. Am J Dermato-pathol 20(1): 79-85.
-
Rozenszajn L, Klajman A, Yaffe D, Efrati P (1966) Jordan’s anomaly in white blood cells. Report Case Blood 28(2): 258-265.
-
Emre S, Unver N, Evans SE, Yüzbaşioğlu A, Gürakan F, et al. (2010) Molecular analysis of Chanarin–Dorfman syndrome (CDS) patients: identification of novel mutations in the ABHD5 gene. Eur J Med Genet 53(3): 141-144.
-
Dorfman ML, Hershko C, Eisenberg S, Sagher F (1974) Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol 110(2): 261-266.
-
Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, et al. (2006) Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin– Dorfman Syndrome. Cell Metab 3(5): 309-319.
- Epithelioid Granuloma; 3cases with Different Clinical Features
- Advancing Representation in Dermatology Clinical Trials: Ethical, Scientific, and Regulatory Imperatives for Inclusion Across all Fitzpatrick Skin Types
- A Case of Atopic Dermatitis with Concurrent Psoriasis Vulgaris: Successful Treatment with Upadacitinib
- Innovation Lifting Eyeshadow: A Synthesis of Makeup and Optical Illusion
- Distinguishing Superficial Actinic Porokeratosis from Actinic Keratosis with UVF Dermoscopy: A Case Report
- High Mobility Group Box 1 (HMGB1) in Cutaneous Inflammation: An Immune Modulator Bridging Cellular Stress, Ferroptosis and Danger Signaling