Angiotensin II Contributes to the Pathophysiology of Heart Failure
Angiotensin is a protein hormone; it is a part of the renin-angiotensin system (RAS), that causes blood vessels to become narrower, helps to maintain blood pressure and fluid balance in the body. It is the primary effector hormone that secreted in response to physiological stimuli. Despite, there is now general agreement that RAS is known to be much more complicated, acting locally within organs or tissue, through both intracrine and autocrine/paracrine mechanisms and systemically, to exert its effects on target organs, including the heart. High secretion of angiotensin II is a common problem resulting in excess fluid retained by the body and, ultimately, raised blood pressure frequency occurs in heart failure where angiotensin is also thought to take part in growing the heart size. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are used in the clinic to avoid these adverse effects. Besides, High levels of Ang II often causes hypertension, renal failure, and cardiac fibrosis. In this review, will be illustrated how Chronic Activation of the renin-angiotensin System Induces heart failure and injures endothelial cells by activation of cellular suicide pathways leading to apoptosis. Its Effects mediated by angiotensin II receptor (AT2) are suggested to include inhibition of cell growth, fetal tissue progression, extracellular matrix modulation, neuronal regeneration, cellular differentiation, apoptosis, and may be vasodilation and left ventricular hypertrophy.
Introduction
In 1898, after renin, angiotensinogen and angiotensin- converting enzyme (ACE) discovering were identified as additional vital components of the classical circulating renin-angiotensin system (RAS) [1]. The precursor of angiotensin is angiotensinogen; it is an α-glycoprotein produced mostly by the liver [2] and released into the systemic circulation then converted to angiotensin I (Ang I) [3]. (Ang I) under effectiveness by renin (Figure 1) Ang I split into Ang II by endothelial-bound ACE in the lungs. Renin explicitly released from the juxtaglomerular cells of the kidneys in response to low pressures. It is converted to angiotensin I by angiotensinogen produced by the liver, which is then converted to angiotensin II by angiotensin- converting enzyme (ACE) in the lungs [4, 5]. The hemodynamic effects of angiotensin II include stimulation of aldosterone production and systemic vasoconstriction and, consequently, fluid retention and increased systemic blood pressures, but it also has several direct cardiac effects. Angiotensin II induces hypertrophy and apoptosis of the cardiomyocytes, and it is the most important regulator of the development of myocardial fibrosis [6]. These effects are central components of cardiac remodelling, which is a maladaptive response causing ventricular dilatation and cardiac dysfunction [7]. On the other hand, the hemodynamic effects of angiotensin II include stimulation of zona glomerulosa cells of the adrenal cortex through its ability in altering renal sodium and water absorption to stimulate the zona glomerulosa cells of the adrenal cortex to synthesise and secrete aldosterone, which influences blood pressure regulation by increasing reabsorption of water and sodium in the kidneys, but it also promotes cardiac fibrosis and endothelial dysfunction [8, 9]. Angiotensin II acts through two types of receptors termed AT1 and AT2. to conduct signals. The activation of the AT2 receptor usually indicates a pathophysiological condition of AT1-mediated action with potentially harmful consequences [10]. This activation may be adaptive in response to acute reductions in cardiac contractile function but has deleterious effects after chronic activity. Angiotensin II promotes peripheral vasoconstriction that increases myocardial oxygen demand. Furthermore, angiotensin II fosters the proliferation of cardiac fibroblasts and the deposition of extracellular matrix resulting in cardiac fibrosis. Also, numerous studies have shown that Ang II signalling in the failing heart associated with increased ROS generation and oxidative stress which causes cardiac hypertrophy, doubled OH• production in the hearts in an AT1R-dependent manner [11].
Methods
Using PubMed, we performed a systematic literature search to identify studies investigating the effects of angiotensin on the heart and how it can cause heart failure conducted between 1998 and 2016. Additionally, all relevant reference lists were screened manually. Studies investigating adults who suffer from heart disease which faces a chronic elevation of Ang II level or aldosterone (ALDO) and the development of heart dysfunction and failure were included in this review.
The Life Cycle of Angiotensin II
Synthesis of angiotensin II-is the last station in renin- angiotensin System (RAS) cascade which begins with altering in sodium/water levels in the blood. First, angiotensin II is an oligopeptide of eight amino acids that causes narrowing in the blood vessels. It maintains blood pressure and fluid balance in the body. It is formed from its original precursor, angiotensinogen, by a series of two enzymatic cleavages [3, 14]. When renal blood flow is lower than usual, the plasma sodium concentration is reduced, in the kidneys prorenin (an intracellular protein) will convert into renin, Renin is an enzyme created via pericytes in the vicinity of the afferent arterioles and juxtaglomerular apparatus cells. It is reserved in intracellular vesicles and rapidly secreted in response to three stimuli: a decrease in blood pressure, a reduction in the sodium concentration delivered to the distal tubules, and activation of the sympathetic nervous system (through β1adrenergic receptors) [3]. Renin itself has no peripheral receptors as well as no direct hemodynamic effects. Directly after secretion into the circulation, then catalyzes cleavage of angiotensinogen which is released from liver by cutting a short 10 amino acid long and peptide off the plasma protein angiotensinogen to angiotensin I. The short peptide angiotensin I is then converted by the removal of 2 amino acids to form the octapeptide angiotensin II, by the angiotensin-converting enzyme (ACE) found in the lung capillaries which is the primary location for (ACE) [13] (Figure 1).
Angiotensin II and aldosterone
Angiotensin II also urges the adrenal cortex to secrete the hormone aldosterone. Hence, it also called renin- angiotensin- aldosterone system (RAAS). Aldosterone triggers and causes increase in the sodium and water reabsorption through the renal tubules, whereas at the same time, to maintain electrolyte balance, causing the excretion of potassium then increases the volume of extracellular fluid in the body, which lead to increases blood pressure [15] (Figure 1).
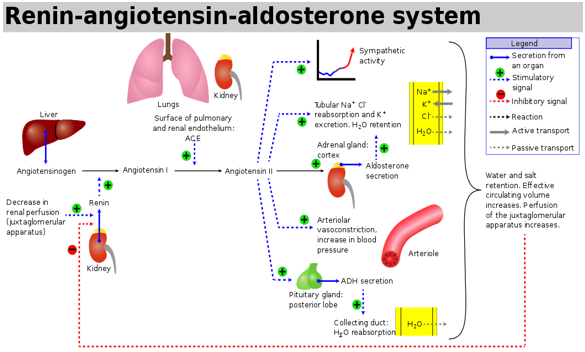
Physiological and pathological effects of angiotensin
• Blood vessels (vascular), causing constriction (narrowing) of the blood vessels then increase blood pressure.
• Nerves (neurological), causing the sensation of thirst, desire increasing for salt, and encouraging the pituitary gland to secrete the anti-diuretic hormone and sympathetic nerves to release noradrenaline.
• Adrenal glands, urging aldosterone production, resulting in the body retaining sodium and losing potassium from the kidneys.
• The kidneys, increasing sodium retention and altering the way the kidneys filter blood which causes rising in the water reabsorption in the kidney which causes an increase in the blood volume and blood pressure.
• On the other hand, in the cardiovascular system, the renin-angiotensin system is a primary component of the physiological and pathological responses. Its primary effector hormone, angiotensin II (ANG II), not only mediates immediate physiological effects of vasoconstriction and blood pressure regulation, but is also implicated in inflammation, endothelial dysfunction, atherosclerosis, hypertension, and congestive heart failure.
• ANG II, via AT1 receptors, carries out its functions, the central signalling mechanisms by which angiotensin affected on cardiovascular physiology and pathology [3, 10, 14, 15, 16].
Angiotensin acts through specific receptors AT1, AT2
Ang II receptors located throughout the heart, blood vessels, kidney, adrenal cortex, lung and brain. Most of the known pathophysiologic effects of Ang II are mediated by AT1R, including vasoconstriction and increased blood pressure, promotion of tissue inflammation and fibrosis, increased oxidative stress, and aldosterone production [10].
Angiotensin II and oxidative stress involved in the heart failure
The RAS is a central feature in the pathophysiology of heart failure acting locally, through both intracrine and autocrine/paracrine mechanisms, as well as systemically to exert its effects on target organs, including the heart. Adaptations in this system occur in response to an inability to meet the demands of vital organ systems. The RAS activated by renal hypoperfusion and sympathetic activation [6, 7]. The principal product of this cascade is angiotensin II, which has multiple system-wide effects that are initially compensatory but subsequently exacerbate and causes heart failure syndrome that attributed to the following: first, Ang II has blood pressure-dependent and blood pressure-independent effects in the heart that cause a crucial role in cardiac remodelling and dysfunction. Cardiac remodelling which results from chronic hypertension or following an acute myocardial infarction which is essential risk factors for heart failure development of and, eventually, death. The renin-angiotensin system (RAS) has previously been established to play a crucial role in the progression of cardiac remodelling, and for protecting the heart from that remodelling then subsequent heart failure the hyperactive RAS should be inhibited [17] (Figure 2).
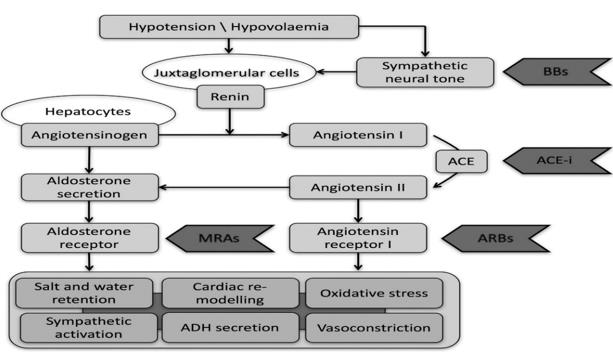
(apoptosis) by activation of cellular suicide pathways. Plenty of studies refer to those endogenous or exogenous elevations in plasma angiotensin II are associated with acute cardiac myocyte necrosis and following by microscopic scarring. As we mentioned above, that Ang II acts as a compensatory factor to recover the deficiency in the heart function [19]. If we look back at the tries and successes of animal evolution, beginning from the peristaltic tube of primitive worms and developed to the four-chamber heart of birds and mammals. It reveals that nature has experimented many different devices for propelling the blood, has constructed various circulatory systems for feeding the organs, and has used several carriers for conveying oxygen. The primitive cardiac tube does not have chambers or valves and had peristaltic movements moving fluid into pericellular spaces, without right vessels or unidirectional flow [20]. Analysing the performances of the heart pumping under this development process may give a clue to the heart adaptation to the constraints urged by the propulsion of blood to appropriate with the organism requirements. Hence, when the heart faces a chronic elevation in plasma Ang II level or aldosterone (ALDO), relative to sodium intake to some extent get the circulatory system back to be similar for the pump that we inherited from our animal ancestors by the independent on the blood vessels constriction to adequate the body needs consequently with a perivascular and interstitial fibrosis of the coronary and systemic circulations then heart failure [21]. Pharmacologic agents which interfere with these effector hormones (e.g., ACE inhibition and ALDO receptor antagonism) protect the myocardium against this pathologic structural remodelling created by the reactive and replacement (reparative) fibrosis [19]. Moreover, Ang II given exogenously to rodents has been shown to result in cellular changes within the myocardium, hypertrophy and eventual fibrosis, like that seen in humans. Some studies demonstrated some of the prospects behind cellular changes in the myocardium that might cause by oxidative stress which is induced by Ang II further, can cause mitochondrial damage and dysfunction [11]. The most important is the split of angiotensinogen AGT by renin was latterly shown to require a conformational change was brought by a formation of a disulphide bridge between two cysteines as a result of oxidation, which refers to the presence of a positive feedback mechanism by which Ang II-induced ROS also, increase Ang II formation [22, 23] (Figure 3).
![Figure 3: Redox modulation of AGT cleavage and formation of a disulfide bridge. Ang I- angiotensin I; AGT- angiotensinogen; (P)RR-(pro)renin receptor. This ongoing ROS production usually counteracted, and an equilibrium oxidative state maintained by SODs and the reduction of H2O2 to H2O by catalase and peroxidases, as well as the actions of nonenzymatic antioxidants and radical scavengers. Many cellular proteins change in conformation and activity due to reduction and oxidation of their thiol groups, and ROS have been shown to play a vital role in many important physiological processes, including host defence and inflammation, cellular signalling, gene expression and regulation of cell growth and death, and oxygen sensing [24].](/fulltextimages/3168/fig_3.jpeg)
Figure 3: Redox modulation of AGT cleavage and formation of a disulfide bridge. Ang I- angiotensin I; AGT- angiotensinogen; (P)RR-(pro)renin receptor. This ongoing ROS production usually counteracted, and an equilibrium oxidative state maintained by SODs and the reduction of H2O2 to H2O by catalase and peroxidases, as well as the actions of nonenzymatic antioxidants and radical scavengers. Many cellular proteins change in conformation and activity due to reduction and oxidation of their thiol groups, and ROS have been shown to play a vital role in many important physiological processes, including host defence and inflammation, cellular signalling, gene expression and regulation of cell growth and death, and oxygen sensing [24].
Heart failure (HF) characterized by activation of the sympathetic nervous and renin-angiotensin-aldosterone systems. This neuroendocrine activation associated with oxidative stress in the myocardium and vasculature. Oxidative stress is an imbalance between the generation and detoxification of reactive oxygen species (ROS) [25, 26]. In patients with HF, oxidative stress occurs in the myocardium [27, 28]. and plasma and correlates with left ventricular (LV) dysfunction [29]. Reactive oxygen species negatively affect the disposition of myocardial calcium (Ca2+), cause arrhythmia, and contribute to cardiac remodelling by inducing hypertrophic signalling, apoptosis, and necrosis [30, 31]. Enzymatic sources for ROS, such as the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), uncoupled nitric oxide (NO) synthase, and mitochondria are all considered relevant sources of ROS in HF, causing vascular and myocardial dysfunction. Importantly, mitochondria amplify ROS derived from NOXs and may thereby function as “redox hubs” in cardiac physiology and disease. As mentioned above, many of the downstream effects of Ang II signalling mediated through ROS generation. Ang II-induced ROS modulate gene expression through transcription factor activation, upregulate growth factors and cytokines, and activate kinases. Oxidative stress has been revealed to be significant in mediating Ang II- induced cardiac hypertrophy, apoptosis, and fibrosis. As well as plays a crucial role in inflammation, and cardiomyocyte contractile dysfunction [32, 33]. Taken together, this is strong evidence which supports that Ang II role in the development of myocardial fibrosis consequently heart failure.
Conclusion
The RAS is a central feature in the pathophysiology of heart failure acting locally, Adaptations in this system occur in response to an inability to meet the demands of vital organ systems. The RAS which is activated by renal hypoperfusion and sympathetic activation. The principal product of this cascade is angiotensin II, which has multiple system-wide effects that are initially compensatory but subsequently exacerbate and causes heart failure syndrome. When the heart faces a chronic elevation in plasma Ang II level or aldosterone (ALDO) relative to sodium intake, both, to some extent get the circulatory system back to be like the pump that we inherited from our animal ancestors through the rely on the constriction of the blood vessels to meet the body needs. Consequently, perivascular and interstitial fibrosis of the coronary and systemic circulations then heart failure. On the other hand, some of the prospects behind cellular changes in the myocardium that might cause by oxidative stress which is induced by Ang II further, can cause mitochondrial damage and dysfunction and negatively affect the disposition of myocardial calcium (Ca2+), which cause arrhythmia, and contribute to cardiac remodelling. Pharmacologic agents which interfere with these effector hormones (e.g., ACE inhibition and ALDO receptor antagonism) protect the myocardium against this pathologic structural remodelling created by the reactive and replacement (reparative) fibrosis.
References
-
Tigerstedt R, Bergman P (1898) Niere and Kreislauf. Scand Arch Physiol 8: 71-223.
-
Correa TD, Takala J, Jakob S (2015) Angiotensin II in septic shock. Crit Care 19: 98.
-
Paul M, Mehr A, Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86(3): 747– 803.
-
Carey RM, Wang ZQ, Siragy HM (2000) Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 35(1): 155-163.
-
McKinley MJ, Johnson AK (2004) The physiological regulation of thirst and fluid intake. News Physiol Sci 19: 1-6.
-
Sciarretta S, Paneni F, Palano F, Chin D, Tocci G, et al. (2009) Role of the renin-angiotensin-aldosterone system and inflammatory processes in the development and progression of diastolic dysfunction. Clin Sci 116(6): 467-477.
-
Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, et al. (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 117(7): 1782-1793.
-
Pacurari M, Kafoury R, Tchounwou PB, Ndebele K (2014) The renin-angiotensin-aldosterone system in vascular inflammation and remodeling. Int J Inflamm pp: 13.
-
Buglioni A, Cannone V, Cataliotti A, Sangaralingham SJ, Heublein, DM, et al. (2015) Circulating aldosterone and natriuretic peptides in the general community: Relationship to cardiorenal and metabolic disease. Hypertension 65(1): 45-53.
-
Putnam K, Shoemaker R, Yiannikouris F, Cassis LA (2012) The renin-angiotensin system: A target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am J Physiol Heart Circ Physiol 302(6): 1219-1230.
-
Hayashi H, Kobara M, Abe M, Tanaka N, Gouda E, et al. (2008) Aldosterone non-genomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertens Res 31(2): 363-375.
-
Chawla LS, Busse LW, Brasha-Mitchell E, Alotaibi Z (2016) The use of angiotensin II in distributive shock. Crit Care 20(1): 137.
-
Orfanos SE, Armaganidis A, Glynos C, Psevdi E, Kaltsas P, et al. (2000) Pulmonary capillary endothelium- bound angiotensin-converting enzyme activity in acute lung injury. Circulation 102(16): 2011-2018.
-
Kopf P, Campbell W (2013) Endothelial metabolism of angiotensin II to angiotensin III, not angiotensin (1– 7), augments the vasorelaxation response in adrenal cortical arteries. Endocrinology 154(12): 4768-4776.
-
Yee AH, Burns JD, Wijdicks EF (2010) Cerebral salt wasting: pathophysiology, diagnosis, and treatment. Neurosurg Clin N Am 21(2): 339-352.
-
Fyhrquist F, Saijonmaa O (2008) Renin-angiotensin system revisited. J Intern Med 264(3): 224-236.
-
Santiago NM, Guimarães PS, Sirvente RA, Oliveira LA, Irigoyen MC, et al. (2010) Lifetime overproduction of circulating Angiotensin-(1-7) attenuates deoxycorticosterone acetate-salt hypertension- induced cardiac dysfunction and remodeling. Hypertension 55(4): 889-896.
-
Ikeda Y1, Nakamura T, Takano H, Kimura H, Obata JE, et al. (2000) Angiotensin II-induced cardiomyocyte hypertrophy and cardiac fibrosis in stroke-prone spontaneously hypertensive rats. J Lab Clin Med 135(4): 353-359.
-
Weber KT, Brilla CG, Campbell SE, Guarda E, Zhou G, et al. (1993) Myocardial fibrosis: role of angiotensin II and aldosterone. Basic Res Cardiol 88(1): 107-124.
-
Boogerd CJJ Moorman AFM, Barnett P (2009) Protein interactions at the heart of cardiac chamber formation. Ann Anat 191(6): 505-517.
-
Bishopric NH (2005) Evolution of the heart from bacteria to man. Ann N Y Acad Sci 1047: 13-29.
-
Mehta PK, Griendling KK (2007) Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292(1): 82-97.
-
Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M (2000) Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol 20(10): 2175-2083.
-
Bedard K, Krause KH (2007) The NOX family of ROS- generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1): 245-313.
-
Gori T, Münzel T (2011) Oxidative stress and endothelial dysfunction: therapeutic implications. Ann Med 43(4): 259-272.
-
Münzel TT, Gori RM, Bruno ST (2010) Is oxidative stress a therapeutic target in cardiovascular disease?. Eur Heart J 31(22): 2741-2748.
-
Maack CT, Kartes H (2003) Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circul 108(13): 1567- 1574.
-
Mollnau H, Oelze M, August M, Wendt M, Daiber A, et al. (2005) Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler Thromb Vasc Biol 25: 2554-2559.
-
Belch JJ, Bridges AB, Scott N, Chopra M (1991) Oxygen free radicals and congestive heart failure. Br Heart J 65: 245-248.
-
Burgoyne JR, Mongue-Din H, Eaton P, Shah AM (2012) Redox signaling in cardiac physiology and pathology. Circ Res 111(8): 1091-1106.
-
Wagner S, Rokita AG, Anderson ME, Maier LS (2013) Redox regulation of sodium and calcium handling. Antioxid Redox Signal 18(9): 1063-1077.
-
Kröller-Schön S, Steven S, Kossmann S, Scholz A, Daub S, et al. (2014) Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid Redox Signal 20(2): 247-266.
-
Maack C, Böhm M (2011) Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J Am Coll Cardiol 58(1): 83-86.
- Acido Labile or Gastro Irritant Apis and Enteric Release in Galenic Practice: An Overview
- A Study on Knowledge, Attitude and Practice of Hand Hygiene among Healthcare Professionals at a Tertiary Care Hospital, India
- Influence of Inoculum Concentration on In Vivo Incubation Period of Emmia lacerata, Pathogenesis and Management of Wilt in Pepper (Capsicum annuum L.)
- Vanilla’s Chemistry
- Marine Anti-Cancer Compounds and Adverse Effects of Global Warming on Oceans: An Overview
- Serological Investigation of Chikungunya Virus Antibody among Malaria-Suspected Febrile Patients in Some Healthcare Facilities in Rivers State