Insights into the Effect of Trehalose and Cyclodextrin Molecules on Dissolution Characteristics of Furosemide Lyophilised Powder
The aim of this work is to enhance the dissolution of furosemide, a hydrophobic drug used in treating hypertension. Solid dispersion via lyophilisation was employed for this purpose. Sugar and oligosaccharide, trehalose and hydroxyl propyl-γ-cyclodextrin (HP-γ-CD), were used as carrier molecules. Complexation of drug molecules with cyclodextrins changes its physical properties, including solubility. Trehalose was selected as it is a hydrophilic molecule as well as it reduces the cellular oxidative stress. Binary solid dispersions at three weight ratios drug to sugar/oligosaccharide were prepared. Solid dispersions were compared with unprocessed drug molecules and the marketed product. Samples were characterized via Fourier transform infrared spectroscopy, differential scanning calorimetry and In vitro drug dissolution. All lyophilized samples showed a significant improvement (p < 0.05) in furosemide dissolution in the aqueous dissolution medium. The study introduces furosemide/HP-γ-CD and furosemide/trehalose freeze dried composites as potential candidate powders to formulate for example fast disintegrating tablets.
Introduction
Poor aqueous solubility of certain drugs is one of the most significant challenges in drug development process. It is one of the key factors that controls dissolution rate, and hence is expected to affect the bioavailability of the therapeutic agent [1]. The oral route of drug administration is the most convenient for drug delivery because of its ease of administration with high patient compliance and is considered to be cost-effective. For efficient drug action after oral administration, the drug particles must be first dissolved in the gastrointestinal fluids prior to being absorbed into the systemic circulation. Incomplete drug absorption of certain candidates from the intestines leads to interruption in drug distribution, reduced bioavailability and therapeutic failures [2]. The oral bioavailability for drugs relies on a few factors, including solubility and dissolution in the gastrointestinal fluids, drug permeability across the gut barriers, and metabolism by enzymes. The most common causes of low oral bioavailability are resulted from poor solubility and/or low permeability of certain drugs [3]. Poorly water-soluble drugs often require high doses to reach therapeutic plasma concentrations after oral administration [4]. According to the Biopharmaceutical Classification
System (BCS), class II drugs have high permeability and low solubility, where the bioavailability of these agents is limited by their dissolution rate. Furosemide (Figure 1A) is a model BCS Class IV hydrophobic agent that suffers incomplete and variable bioavailability due to its limited aqueous solubility and permeability.
Many approaches have been tried to improve the dissolution of poorly water soluble therapeutics such as using liquisolid techniques [5, 6, 7], solid dispersions [8], nanosuspensions [9], liposomes [10] and complexation with cyclodextrins [11].
Cyclodextrins (CDs) are oligosaccharides arranged in a cyclic form with the hydrophilic surface oriented outwards. This gives CDs their unique lipophilic central cavity that can accommodate lipophilic molecules via complex formation. This complex formation can modify physical properties of lipophilic molecules, including aqueous drug solubility. Usually, substituted CDs were prepared to overcome the limited solubility of natural CD [12]. hydroxyl-propyl-γ- cyclodextrin (HP-γ-CD) is one such example of substituted CD (Figure 1B). Based on the unique ability of CDs to a accommodate guest molecules in their internal cavity, they have been successfully used to increase solubility of hydrophobic drugs [13].
Trehalose is widely used as a carrier to increase protein stability, based on its chemical structure (Figure 1C). Recently it was reported that oxidative stress is a key factor in the pathogenesis of hypertension [14]. Exposure of cells to reactive oxygen species, such as hydrogen peroxide, can cause oxidative damage to amino acids in the cellular proteins, and trehalose accumulation was found to reduce such damage [15]. Therefore, trehalose was selected as carrier to furosemide based on many reasons. First, being hydrophilic sugar it is expected to increase wettability and consequently solubility of lipophilic drugs. Other sugars, such as sucralose, were investigated as potential solubility modifier with reported success [16]. Secondary, due to its protection against oxidative stress that may augment furosemide effect in controlling hypertension.
In this study, the aim was to improve furosemide dissolution rate via hydrophilic sugars employing solid dispersion technique, namely freeze drying. Sugars are known to enhance dissolution of poorly water-soluble drugs [17]. However due to the sparse literature, HP-γ-CD and Trehalose were chosen to confirm the suitability of those specific carriers to enhance hydrophobic drug dissolution.
Materials and Methods
Materials
Furosemide, 2-hydroxypropyl-γ-cyclodextrin, trehalose were obtained from Sigma-Aldrich (Dorset, UK). Other chemicals were of analytical grade and used as obtained.
Methods
Preparation of furosemide freeze dried formulations: Solid dispersion formulations of furosemide were prepared employing freeze drying technique. Binary solid dispersion of the drug and the hydrophilic excipients were prepared at different weight ratios according to compositions shown in Table 1.
| Formula* | Furosemide (gm) | Cyclodextrin (gm) | Trehalose (gm) | Q10 | Q90 |
|---|---|---|---|---|---|
| control | 13.8±0.7 | 42.2±0.4 | |||
| FCD1 | 1 | 1 | 84.5±0.8 | 97.9±0.8 | |
| FCD2 | 1 | 2 | 94.8±0.4 | 98.9±0.2 | |
| FCD3 | 1 | 5 | 95.1±0.7 | 99.2±0.2 | |
| FT1 | 1 | 1 | 64.5±5.2 | 93.8±0.3 | |
| FT2 | 1 | 2 | 39.3±4.1 | 91.1±3.3 | |
| FT3 | 1 | 5 | 55.4±0.1 | 99.9±0.3 | |
| Marketed product | - | - | - | 63.0±0.4 | 90±0.7 |
Table 1: Compositions of solid dispersions of furosemide with the chosen hydrophilic excipients * FCD is formulae with cyclodextr
Table 1: Compositions of solid dispersions of furosemide with the chosen hydrophilic excipients * FCD is formulae with cyclodextrin *FT is formulae with trehalose Drug and carrier were dissolved in 1:1 ethanol: water ratio (this ratio was chosen as it has resulted in a complete solubilisation of the powder). The obtained solutions were subjected to freezing at −85◦C for 24 hours before subjecting to lyophilization (VirTis freeze drier, BioPharma, Boston, MA, USA). Shelf temperature was kept at 20◦C under pressure of 18mBar.
Characterization of furosemide formulations • Fourier Transform Infra-Red Spectroscopy (FT-IR) Fourier Transform Infra-Red spectroscopy (FT-IR) was conducted for the unprocessed furosemide, pure hydrophilic excipients and their binary solid dispersion formulations. Small amounts of all samples were used. The spectra were measured over the range between 4000–500 cm−1, and the resolution used was 4 cm−1 using FTIR spectrophotometer (SHIMADZU, Buckinghamshire, UK).
• Differential Scanning Calorimetry (DSC) The thermal behavior of unprocessed furosemide, pure hydrophilic excipients and their binary solid dispersions were investigated using DSC (Q 1000 TA, TA Instruments, Ghent, Belgium). Samples, each weighing about 3 mg, were loaded into aluminum pans and hermetically sealed, using empty pan as reference. The test and reference pans were heated simultaneously from 0 to 300oC at a heating rate of 10oC/minute.
• Dissolution Studies In vitro drug dissolution studies were performed on the lyophilized formulations and unprocessed furosemide drug powders (20mg) as control. The dissolution tests were performed using USP dissolution apparatus type II (Caleva Ltd., Dorset, UK). In this method, 900 mL distilled water was used as a dissolution medium (as the aim of the study was to test drug aqueous dissolution characteristics in water as a general aqueous medium). The samples (equivalent to
20mg of the drug) were loaded into the dissolution vessels. Temperature was kept at 37 ± 1 °C and the medium was stirred at a paddle speed of 100 rpm (13). Ten milliliters of the samples were collected at intervals of 5, 10, 15, 20, 25, 30, 45, 60 and 90 min and were replaced by 10 mL to maintain a constant volume. Then the collected samples were analysed spectrophotometrically at 274 nm using M501 single beam scanning ultra-violet spectrophotometer (Spectronic Tudor, Leeds, UK). All the dissolution tests were performed in triplicates. For comparison, the marketed product (Furosemide 20mg, Bristol LTD) was used to evaluate our formulations.
Stability studies In this study, the accelerated stability study was performed to study the effect of ageing on furosemide in selected formulations. Samples were stored in stability cabinet with conditions of 40 ± 2°C/75% ± 5% RH for 6 months, and their properties were examined by conducting dissolution studies.
Statistical analysis All data obtained from the evaluation of solid dispersions were subjected to the appropriate statistical analysis, using student t-test, to determine their significances. The significance level used was 0.05, and results are regarded as significant when P-value is less than 0.05.
Results and Discussion
Characterisation of Furosemide Freeze Drier Formulations
Differential scanning calorimetry (DSC): Thermal pattern of furosemide before and after processing with different excipients were investigated. The collected thermograms are shown in Figure 2.
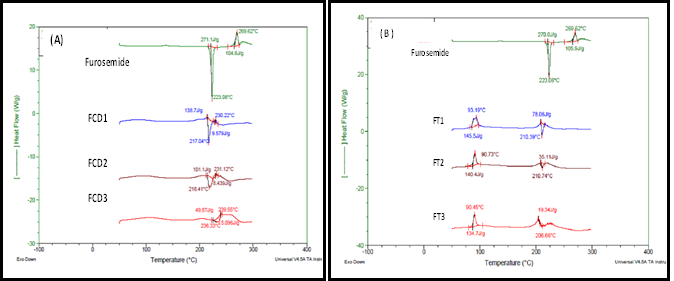
DSC was used for the investigation of any interaction between the drug and the used excipients. The DSC thermogram for unprocessed furosemide showed two peaks, one is a sharp exothermic peak at 223.08°C with high enthalpy of 270.1 J/g corresponds to its melting transition (Tm) and reflects its crystalline nature. A second endothermic peak was noticed at about 269.62°C that may correspond to its degradation. This thermogram is similar to published data for the same drug [18]. Any change in the position or the intensity of these two peaks could be taken as changes in the physical or chemical characteristics of the drug.
Lyophilization of furosemide with cyclodextrin resulted in shifting of the main transition peak of furosemide to lower Tm with the peak becoming broader with reduced sharpness. This accompanied by reduction in the melting enthalpy. Increasing cyclodextrin concentration in FCD3 (1:5 drug to cyclodextrin, respectively) resulted in complete abolish of the drug main transition peak indicating possible complete drug amorphousization arising from the fact that drug is molecularly entrapped in the cavity of CD. It worth noting that the melting transition is immediately followed by endothermic event. This could be due to the decomposition of the newly formed complex. These finding were previously reported by other investigators and were similarly explained [19].
For furosemide/trehalose freeze dried samples, the thermograms showed two thermal events. The first is an endothermic peak at about 93ͦ°C that corresponds to melting transition of trehalose [20]. This indicates that trehalose was in the dehydrate form [21]. The second is for the melting of furosemide that was shifted to lower temperature (210°C). As the concentration of trehalose increases, the transition peak of the drug becomes smaller and more broader with Tm of 206.6°C. This can indicate reduced drug crystalinity or weakening of the crystalline structure of the drug with possible development of amorphous form. At the highest ratio of trehalose, the thermal event showed further broadening with peak sharpening markedly reduced. This could indicate transformation of appreciable amount of the drug to the amorphous state. Fourier transform infra-red spectroscopy (FT-IR) results: The structural features of furosemide were identified using FTIR before and after processing. The spectra (data not shown) reflected the characteristic peaks for unprocessed drug and different formulations. Pure furosemide showed a characteristic absorption bands at 3350 cm−1 which is assigned to the NH2 stretching vibration of -NHCH2, another band at 3260 cm−1 due to stretching vibration of SO2NH2 and band 1660 cm−1 assigned to the bending vibration of the amino group. The band due to the asymmetric stretching vibration of the carbonyl group is noticed at 1560 cm−1 and the 1318 cm−1 band is for the asymmetric stretching vibration of the sulfonyl group in furosemide structure. The drug spectrum is similar to the previously reported drug spectrum [18, 22].
Regarding the spectra of different freeze dried formulations using different excipients, the main characteristic peaks of furosemide can be detected (data not shown). This indicates that there is no interaction between the hydrophilic excipients and the drug.
In Vitro Drug Dissolution Testing
The dissolution profiles of furosemide released from different formulations are presented in Figure 2. The dissolution parameters taken as percentage amount released after 10 minutes (Q10) and total percentage drug released after 90 minutes (Q90) are presented in Table 1. The dissolution pattern of unprocessed furosemide showed slow and incomplete drug dissolution with Q10 of only 13% with an overall release of 42% of the loaded dose at the end of the dissolution study. This dissolution behavior can be explained based on the poor drug solubility.
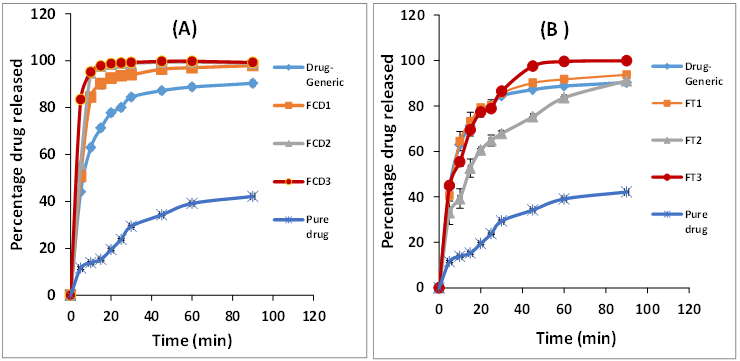
Freeze dried formulations using HP-γ-CD significantly (P < 0.05) increased furosemide dissolution (Figure 3). The extent of enhancement correlated to the proportion of HP-γ- CD in the solid dispersion. At 1:1 ratio (formula FCD1), about 50% of the loaded drug was liberated after 5 minutes that was increased to 83% after 10 minutes (Q10) giving rise to about 6-fold enhancement in drug dissolution, compared to unprocessed. Increasing HP-γ-CD proportion to 1:2 slightly increased dissolution parameters relative to FCD1. However, for FCD3 (1:5 drug: HP-γ-CD) there was a prompt release of 83 and 94% after 5 and 10 minutes, respectively. Such high initial drug release is very advantageous when fast disintegrating solid dosage form is required. The improved dissolution could be due to reduced drug crystalline nature as the drug is now molecularly dispersed in the cavities of HP-γ-CD molecules. This suggestion is supported by the DSC data. This entrapment takes place by hydrogen bond formation between the hydroxyl groups of CDs and the carboxylic and/or amino groups of furosemide. This would change the physicochemical property of the entrapped drug as reflected by improved dissolution behavior. Inclusion complexation have been long used as solubiliser and succeeded in enhancing the solubility of many poorly soluble drugs by their unique ability to form inclusion complexes with the hydrophobic drugs [20, 23, 24, 25].
For trehalose binary system, the dissolution profiles are shown in Figure 3B and the extracted dissolution parameters are in Table 1. There was a significant (P < 0.05) increased in furosemide dissolution relative to unprocessed form (Figure 3B & Table 1). At 1:1 ratio (FT1), about 64% of the drug was liberated after 10 minutes with a total release of 93% (Q90). Though this result less than that obtained from the same ratio using HP-γ-CD, but were similar to that of the marketed drug product (P < 0.05). It worth noting that, increasing trehalose content to 1:2 (FT2) and 1:5 (FT3), didn’t add significantly (P < 0.05) to drug dissolution rate. Therefore, 1:1 can be considered as the optimum weight ratio. The improved drug dissolution can be explained based on the formation of amorphous form or weakening of the crystalline structure of the drug crystals. This proposition can be supported by the DSC data. This, together with enhanced wettability by the close proximity with the hydrophilic trehalose molecules, can explain the improved dissolution parameters. Similar finding was reported where HP-γ-CD was superior to trehalose in enhancing the dissolution of Bendroflumethiazide [20].
With respect to marketed drug product, an initial release of 63% of loaded dose was noted followed by gradual drug dissolution (Figure 3). Compared to the solid dispersions, freeze dried formulations showed similar (FT1) or even better (FC1-3) dissolution behavior. This would rationalize the suitability of the adopted procedure as well as the selected carriers.
To investigate the stability of the prepared formulation, formulations FCD3 and FT1 were stored at high temperature (40 ± 2°C) and relative humidity of 75% ± 5% for 6 months. After this period, dissolution studies were conducted. This quality test was selected for evaluation because dissolution pattern is critical as the aim of the prepared solid dispersion, employing lyophilization, was to produce formulations with fast drug dissolution suitable for example for preparation of oral dispersible tablets with subsequent rapid drug release.
All tested samples containing either HP-γ-CD or trehalose were stable as there were no significant difference (P< 0.05) in release parameters compared to the freshly prepared samples. Hence, those excipients are promising to be applied with other hydrophobic drugs.
Conclusion
The dissolution rate is the determining step for oral absorption of the poorly water-soluble drugs. Because of poor dissolution and solubility of many drugs, their bioavailability is highly affected. Hence, dissolution and solubility enhancement became necessary task. Hydroxyl propyl-γ-cyclodextrin and Trehalose molecules are promising excipients for enhancing drug dissolution when subjected to lyophilization. Both excipients successfully enhanced the dissolution and solubility of the hydrophobic furosemide. The obtained solid dispersion was stable as reflected from similar dissolution parameters compared to freshly prepared samples. The optimum solid dispersion formulations can be formulated in the future into oral dispersible tablets. This tablet type is suitable for elderly patients whom are the most susceptible group for chronic diseases, including hypertension.
References
-
Savjani KT, Gajjar AK, Savjani JK (2020) Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm pp: 1-10.
-
Xu J, Luo KQ (2014) Enhancing the solubility and bioavailability of isoflavone by particle size reduction using a supercritical carbon dioxide-based precipitation process. Chem Eng Res Design 92(11): 2542-2549.
-
Vasconcelos T, Sarmento B, Costa P (2007) Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Disc Today 12(23-24): 1068-1075.
-
Bremmell KE, Prestidge CA (2019) Enhancing oral bioavailability of poorly soluble drugs with mesoporous silica based systems: opportunities and challenges. Drug Dev Ind Pharm 45(3): 349-358.
-
Chella N, Shastri N, Tadikonda RR (2012) Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm Sinica B 2(5): 502-508.
-
Elkordy AA, Tan XN, Essa EA (2013) Spironolactone release from liquisolid formulations prepared with CapryolTM 90, Solutol® HS-15 and Kollicoat® SR 30 D as non-volatile liquid vehicles. Eur J Pharm Biopharm 83(2): 203-223.
-
Fahmy RH, Kassem MA (2008) Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur J Pharm Biopharm 69(3): 993-1003.
-
Essa EA, Balata GF (2012) Preparation and characterization of domperidone solid dispersion. Pak J Pharm Sci 25(4): 783-791.
-
Cerdeira AM, Mazzotti M, Gander B (2010) Miconazole nanosuspensions: Influence of formulation variables on particle size reduction and physical stability. Int J Pharm 396(1-2): 210-218.
-
Uhl P, Pantze S, Storck P, Parmentier J, Witzigmann D, et al. (2017) Oral delivery of vancomycin by tetraether lipid liposomes. Eur J Pharm Sci 108: 111-118.
-
Loh GOK, Tan YTF, Peh K (2016) Enhancement of norfloxacin solubility via inclusion complexation with β-cyclodextrin and its derivative hydroxypropyl-β- cyclodextrin. Asian J Pharm Sci 11(4): 536-546.
-
Saokham P, Muankaew C, Jansook P, Loftsson T (2018) Solubility of Cyclodextrins and Drug/Cyclodextrin Complexes. Molecules 23(5): 1161.
-
Kim D, Lee S, Pyo Y, Tran P, Park JS (2020) Solubility enhancement and application of cyclodextrins in local drug delivery. J Pharm Investig 50: 17-27.
-
Rodrigo R, González J, Paoletto F (2011) The role of oxidative stress in the pathophysiology of hypertension. Hypertens Res 34(4): 431-440.
-
Benaroudj N, Lee DH, Goldberg AL (2001) Trehalose Accumulation during Cellular Stress Protects Cells and Cellular Proteins from Damage by Oxygen Radicals. J Biolog Chem 276(26): 24261-24267.
-
Essa EA, Elbasuony AR, Abdelaziz AE, El Maghraby GM (2019) Co-crystallization for enhanced dissolution rate of bicalutamide: preparation and evaluation of rapidly disintegrating tablets. Drug Dev Ind Pharm 45(8): 1215- 1223.
-
Blagden N, de Matas M, Gavan PT, York P (2007) Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv Drug Deliv Rev 59(7): 617-630.
-
Akinlade B, Elkordy AA, Essa E, El hagar S (2010) Liquisolid Systems to Improve the Dissolution of Furosemide. Sci Pharm 78(2): 325-344.
-
Kreaz RM, Abu-Eida EY, Eros I, Kata M (1999) Freeze- Dried Complexes of Furosemide with β-Cyclodextrin Derivatives. J Incl Phen Macrocyc Chem 34: 39-48.
-
Saleh A, McGarry K, Chaw CS, Elkordy AA (2018) Feasibility of Using Gluconolactone, trehalose and hydroxy-propyl gamma cyclodextrin to enhance bendroflumethiazide dissolution using lyophilisation and physical mixing techniques. Pharmaceutics 10(1): 22.
-
Jain NK, Roy I (2009) Effect of trehalose on protein structure. Protein Sci 18(1): 24-36.
-
Shin S, Kim J (2003) Physicochemical characterization of solid dispersion of furosemide with TPGS. Int J Pharm 251(1-2): 79-84.
-
Brewster ME, Loftsson T (2007) Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev 59(7): 645-666.
-
Lucio D, Irache JM, Font M, Martínez-Ohárriz MC (2017) Supramolecular structure of glibenclamide and β-cyclodextrins complexes. Int J Pharm 530(1-2): 377- 386.
-
Stoyanova K, Vinarov Z, Tcholakova S (2016) Improving Ibuprofen solubility by surfactant-facilitated self- assembly into mixed micelles. J Drug Deliv Sci Tech 36: 208-215.
- Acido Labile or Gastro Irritant Apis and Enteric Release in Galenic Practice: An Overview
- A Study on Knowledge, Attitude and Practice of Hand Hygiene among Healthcare Professionals at a Tertiary Care Hospital, India
- Influence of Inoculum Concentration on In Vivo Incubation Period of Emmia lacerata, Pathogenesis and Management of Wilt in Pepper (Capsicum annuum L.)
- Vanilla’s Chemistry
- Marine Anti-Cancer Compounds and Adverse Effects of Global Warming on Oceans: An Overview
- Serological Investigation of Chikungunya Virus Antibody among Malaria-Suspected Febrile Patients in Some Healthcare Facilities in Rivers State