Advancements in Analytical Development and Validation: A Comprehensive Review in Pharmaceutical Sciences
The pharmaceutical industry is dedicated to consistently producing high-quality products at an optimal cost, necessitating the development of robust methodologies across various stages from formulation to commercialization. This review article delves into the critical aspects of designing and validating processes integral to pharmaceutical drug development. Emphasizing the importance of reliable analytical techniques, the review underscores their role in quality assessment of pharmaceutical samples. A cornerstone of pharmaceutical manufacturing is adherence to Good Manufacturing Practice (GMP) regulations, which mandates comprehensive documentation and validation of processes. From raw material procurement to final batch production, validation ensures adherence to quality standards throughout the manufacturing chain. The article meticulously examines the creation and validation of analytical methods, focusing on parameters including accuracy, specificity, and precision, limit of detection (LOD), limit of quantitation (LOQ), ruggedness, robustness, and system suitability testing. These validation parameters are instrumental in routine and stability analyses, guaranteeing the reliability and quality of pharmaceutical products. By elucidating the intricate nuances of validation methodologies, this review contributes to the efficient operation of pharmaceutical companies and the continual advancement of drug development practices.
Introduction
Analytical chemistry is the branch of chemistry dedicated to the qualitative and quantitative study of the components within substances, samples, or mixtures. This discipline encompasses two primary types of analysis:
qualitative analysis and quantitative analysis. Qualitative analysis involves the identification of the components or analytes within a mixture or sample, while quantitative analysis involves the measurement of the amounts of these components or analytes.
Analytical data are indispensable across a range of scientific disciplines, including chemistry, biology, archaeology, and astronomy, as well as in clinical diagnostics. The applications of analytical chemistry are extensive, encompassing the monitoring and control of pollutants, clinical and biological research, geological testing, fundamental and applied scientific research, and quality control within industrial sectors. This broad applicability highlights the crucial role of analytical chemistry in advancing scientific knowledge and ensuring the integrity of various processes and products [1].
Analytical Method
Analytical method refers to the employment of a specific technique and precise step-by-step instructions in the qualitative, quantitative, or structural analysis of a sample for one or more analytes [2]. Analytical Methods are mainly classified into 2 types: Classical method & Instrumental method. The classical method refers to a method in which the signal is proportionate to the absolute amount of analyte. The instrumental method refers to a procedure in which the signal is proportionate to the concentration of analytes.
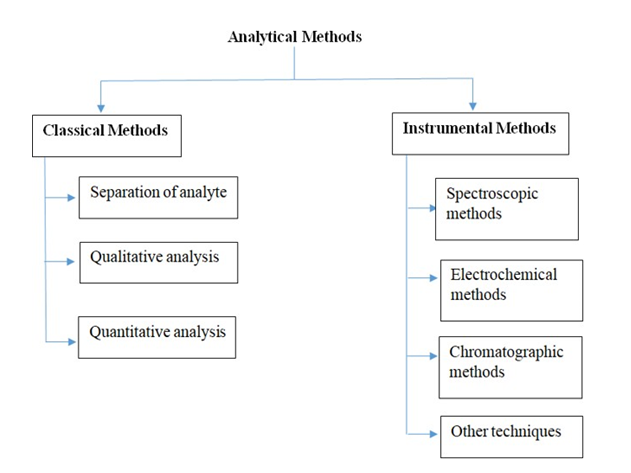
Classical analytical methods are categorized into three primary types: (a) analyte separation, (b) qualitative analysis, and (c) quantitative analysis. Analyte separation techniques include processes such as extraction, distillation, precipitation, and filtration. Qualitative analysis encompasses the determination of physical and chemical properties, such as boiling point, freezing point, color, odor, density, reactivity, and refractive index. Quantitative analysis is exemplified by methods like gravimetric analysis and volumetric analysis.
Instrumental analytical methods are broadly divided into four basic categories: (a) spectroscopic methods, (b) electrochemical methods, (c) chromatographic procedures, and (d) miscellaneous techniques. Spectroscopic methods include UV-visible spectroscopy, infrared spectroscopy, Raman spectroscopy, atomic absorption spectroscopy, atomic emission spectroscopy, X-ray spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy. Electrochemical methods comprise potentiometry, coulometry, and voltammetry. Chromatographic methods encompass column chromatography, paper chromatography, thin-layer chromatography, high-performance liquid chromatography (HPLC), and gas chromatography (GC), along with advanced techniques such as liquid chromatography- mass spectrometry (LC-MS), gas chromatography-mass spectrometry (GC-MS), tandem mass spectrometry (LC- MS-MS, GC-MS-MS), and combined techniques like LC-NMR and GC-NMR. Other instrumental approaches include X-ray methods, radioactivity measurements, mass spectrometry, optical methods (e.g., refractometry, optical rotation), and thermal analysis techniques (e.g., thermogravimetry, differential thermal analysis, and differential scanning calorimetry) [3].
Introduction to Spectroscopy: Spectroscopy is the scientific study of the interaction between electromagnetic radiation and matter, encompassing both the absorption and emission of radiation by substances. Spectroscopy is broadly categorized into two forms: absorption spectroscopy and emission spectroscopy. Absorption spectroscopy, which includes techniques such as UV-visible spectroscopy, infrared (IR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, microwave spectroscopy, and radio wave spectroscopy, focuses on the electromagnetic energy absorbed by a material, producing spectra. Emission spectroscopy, encompassing methods such as flame photometry and fluorimetry, examines the electromagnetic radiation emitted by a sample, also in the form of spectra.
Spectroscopy is a powerful tool for investigating atomic and molecular structures and is widely used for the analysis of various substances. Atomic spectroscopy specifically studies the interactions between electromagnetic radiation and atoms, highlighting energy transitions at the atomic level, as seen in techniques like atomic absorption spectroscopy and flame photometry. Molecular spectroscopy, on the other hand, investigates the interactions between electromagnetic radiation and molecules, focusing on energy changes at the molecular level, as exemplified by ultraviolet (UV) and infrared (IR) spectroscopy.
UV-Vis Spectroscopy: UV-visible spectroscopy quantifies the absorption of light across the ultraviolet (UV) and visible (VIS) regions of the electromagnetic spectrum. This absorption spectroscopy utilizes electromagnetic radiation ranging from 200nm to 800nm, subdivided into ultraviolet (UV, 200-400nm) and visible (VIS, 400-800nm) regions. The principle underlying UV-visible spectroscopy is the absorption of ultraviolet or visible light by a sample or chemical compound, leading to the generation of distinct spectra. When a molecule absorbs UV radiation, its electrons are excited and transition from a lower to a higher electronic energy level, whereas the reverse transition results in ultraviolet emission spectra.
Common solvents used in UV spectroscopy include water, methanol, ethanol, ether, chloroform, carbon tetrachloride, cyclohexane, and dichloroethane. The applications of UV spectroscopy are diverse, encompassing the detection of functional groups, assessment of conjugation, identification of geometrical isomers, and impurity detection [3].
Instrumentation of UV-Visible spectroscopy: Radiation sources: Tungsten lamp, hydrogen discharge lamp, deuterium lamp, xenon discharge lamp, and mercury arc are the most often utilized radiation sources.

- Wavelength Selector- The monochromator is used to disperse radiation based on wavelength. A monochromator’s essential components are an input slit, a dispersing element, and an exit slit.
- Sample Cell- Cells or cuvettes are sample containers used in UV-Visible spectroscopy to hold liquid samples. Quartz is used to make cuvettes.
- Photodetector- The most common UV spectrophotometer detectors are barrier layer cells, photocells, and photomultiplier tubes.
- Readout Device- The detector’s output is amplified and then shown on a readout device [4].
Introduction to Chromatography: Chromatography is a physicochemical process for separating substances from mixtures. Chromatography is a technique for separating a mixture of chemicals into individual components by passing them through two phases, a stationary phase and a mobile phase.
Chromatography is classified as follows:
- Based on interaction of solute to stationary phase • Adsorption chromatography • Partition chromatography • Ion exchange chromatography • Molecular exclusion chromatography
- Based on chromatographic bed shape • Column chromatography • Planar chromatography • Paper chromatography • Thin layer chromatography • Displacement chromatography
- Techniques by physical state of mobile phase • Gas chromatography • Liquid chromatography • Affinity chromatography [5]
HPLC: High-performance liquid chromatography (HPLC), also known as high-pressure liquid chromatography, is a technique utilized to separate, identify, and quantify components in liquid-soluble materials. The core principle of liquid chromatography is adsorption. HPLC operates with a liquid mobile phase, where the sample, in the form of a liquid solution, is introduced into a column containing a porous stationary phase. A pump applies high pressure to propel the sample through the column with the mobile phase. The components of the sample migrate based on their affinity for the stationary phase; those with higher affinity move more slowly, while those with lower affinity move more rapidly, resulting in separation. Common solvents used in HPLC include n-hexane, methylene chloride, chloroform, methyl-t-butyl ether, tetrahydrofuran (THF), isopropanol (IPA), acetonitrile (MeCN or CAN), methanol (MeOH), and water. Key chromatographic parameters in HPLC include efficiency (number of theoretical plates), retention factor, selectivity, resolution, and pressure. HPLC is widely applied for chemical separation, purification, and identification. Additionally, it is employed in various fields such as pharmaceutical, environmental, forensic, clinical, culinary, and flavor analysis [6].
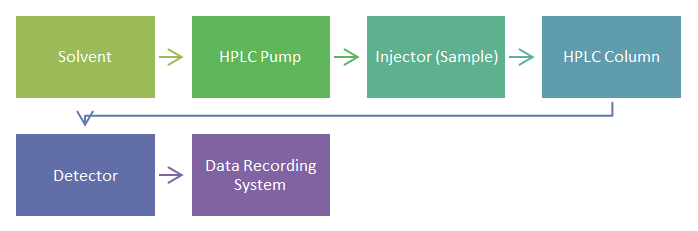
Instrumentation of HPLC: Components of the HPLC system
- Solvent reservoir, mixing system and degassing system
- High pressure pump
- Sample injector
- Column
- Detector
- Data recording system Reservoir for solvent, mixing system, and degassing system: The solvent (mobile phase) is stored in the solvent reservoir. These are containers made of glass or stainless steel that will not discolor. Glass bottles are the most popular type of solvent reservoir. Aside from delivering mobile phase, the pump must also combine solvents with great accuracy and precision. Low pressure mixing and high-pressure mixing are the two types of mixing units. The degassing system eliminates air bubbles that have become captured in the solution. Ultra sonication and filtration are two degassing procedures used [7]. High pressure pump: A pump’s function is to pressurize a liquid and provide a particular flow rate. A milliliter per minute (ml/min) is the unit used to measure flow rate. A flow rate of 1-2 ml/min is typical. The range of a pump’s pressure is 6000-9000 psi (400-600 bars). Constant pressure pumps, syringe pumps, and reciprocating piston pumps are the three most popular types of pumps [8]. Sample injector: A sample injector is used to insert a liquid sample into the mobile phase. Between the pump and the column is a sample valve. The sample can be injected into the continuously flowing mobile phase stream that transports the sample to the HPLC column by an injector (auto sampler). Sample quantities typically range from 5 to 20microliters (l). There are two different types of injectors: manual and automated [8]. Column: The real separation of components happens in the column. Stainless steel makes up the column. Its inside diameter is 2.46cm, and it is 5 to 25cm long [6]. Detector: The detector may turn raw data into an electrical signal by identifying each component that elutes from the column. Specific detectors and bulk property detectors are the two types of detectors that are employed. The UV-VIS detector, photo diode array detector, fluorescence detector, and mass spectrometric detector are examples of particular detectors. Refractive index detectors, electrochemical detectors, and light scattering detectors are examples of bulk property detectors [8]. The Data Recording system: The output is represented as a succession of peaks, and the area beneath each peak can be determined by the computer connected to the display automatically [9].
Developing a New Analytical Method: Criteria
Drug analysis is essential for the accurate identification of pharmaceutical products. There is often a delay between a drug’s market release and its inclusion in pharmacopoeias. This lag is due to several factors: uncertainties regarding the prolonged and expanded use of these therapies, emerging reports of novel hazards, patient resistance, and the introduction of superior drugs by competitors. Consequently, pharmacopoeias may not always have established standards and analytical procedures for these new drugs, necessitating the development of novel analytical techniques. In summary, the need for new drug analysis methods is driven by these factors [4].
It is conceivable that certain pharmaceutical compounds or combinations thereof may lack approval from pharmacopoeias. This scenario could arise due to limitations imposed by patent laws, resulting in a dearth of documented analytical methodologies in the scientific literature. Furthermore, formulations containing these compounds may lack established analytical techniques, particularly regarding excipient components. Challenges may also emerge in devising analytical strategies for assessing the concentration of such compounds in biological fluids, exacerbating the analytical complexity. Current methodologies may necessitate expensive solvents and reagents, along with labor-intensive extraction and separation protocols that may compromise reliability. The sequential steps in method development commence with meticulous documentation, underscoring the imperative of establishing comprehensive record-keeping systems to catalog research findings. All experimental data generated therein must be systematically recorded in either physical laboratory notebooks or electronic databases [4].
Fundamental Standards for Developing New Drug Analysis Techniques:
- The medicine or drug combination might not be recognized by any pharmacopoeias.
- Because of patent laws, a proper analysis technique for the medicine may not be published in the literature.
- Because the formulation excipients interact with the analysis of the drug, analytical procedures might not be available for it.
- The medication may not be quantifiable using analytical procedures in bodily fluids.
- It’s possible that there aren’t any analytical techniques for a drug when it’s taken with other medications.
- Expensive chemicals and solvents might be required by the current analytical techniques.
It could also include laborious extraction and separation techniques, some of which might not be trustworthy [2].
Method Validation
Definition- Analytical method validation is defined as “documented evidence that offers a high level of assurance that a particular process will consistently produce a product meeting its pre-determined specifications and quality attributes” [10].
Parameters of analytical method validation: a) Accuracy b) Precision
- Repeatability
- Intermediate Precision
- Reproducibility c) Specificity d) Detection Limit e) Quantitation limit f) Linearity g) Range h) Stability i) Robustness [10]
Accuracy: The definition of accuracy of an analytical method is “The proximity of the check results obtained using that method to the appropriate price.” Throughout its variety, this correctness must be installed. Any of the following methods can be used to assess the correctness of an analytical procedure. Using a sample of known awareness to analyze and contrast the measured price with the “genuine” price. However, a sample that has been thoroughly studied (such as a well-known reference) should be used. A placebo (product matrix) recovery technique is spiked. On this procedure, a sample that contains all other elements save the energetic(s) is provided to components clean, the resulting combination is evaluated, and the results obtained are compared to the anticipated result.
Approach to general addition In this procedure, a sample is analyzed, a known amount of pure energetic constituent is added, and the pattern is analyzed once more. The difference between the two assays’ impact is in comparison to the expected result. Restoration is described as a percentage of the found result to the intended result in both procedures (spiked -placebo restoration and preferred addition strategy) [11].
The accuracy of a procedure can also varies within the range of possible assay values, so it should be tested at several distinct fortification phases. The precision should cover at least three concentrations (80, 100, and 120%) within the intended range [12].
Accuracy will also be determined by comparing examine outcomes to those acquired using another proven check procedure. Dosage shape assays are typically accurate to within 3-5% of the true value. The ICH files indicate that accuracy be evaluated using a minimum of nine determinations spread throughout at least 3 awareness levels, overlapping the needed range (i.e., 3 concentrations and 3 duplicated willpowers for each attention) [11, 12].
Precision: It expresses the degree of proximity of settlement (diploma of scatter) between a series of measurements acquired from multiple samplings of the same homogeneous sample under the specified parameters. Precision can be divided into three categories: repeatability, intermediate precision, and reproducibility. Repeatability is often referred to as intra-assay precision. It is a measure of the precision of evaluation in a single laboratory by a single operator utilizing a single piece of system in a surprisingly short period of time. It is the degree of impact settling while experimental settings are kept as consistent as possible and is given as RSD of reflects values. ICH suggests at least nine determinations covering the desired variety for the technique (e.g., three concentrations/3 replicates as in the accuracy test), or at least six determinations at 100% of the check concentration for evaluation of repeatability, which should be stated as popular deviation, relative general deviation (coefficient of variation), or self-belief in programming.
Intermediate precision is defined by ICH as long- term variability of the dimension method and is obtained by comparing the results of a method run inside a single laboratory over a number of weeks. It is also known as intraday precision. Reproducibility expresses the precision of evaluating the same pattern by different analysts in different laboratories under different operational and environmental variables that may differ but remain within the technique’s specific parameters.
Specificity: Specificity is the ability to evaluate an analyte unequivocally in the presence of additives that are expected to be present. Impurities, degradants, matrix, and so on are common examples. The lack of specificity of an individual analytical technique may be compensated for by another helping analytical method.
The ICH classifies the term specificity into two distinct groups. Identity: to ensure that an analyte can be identified. Impurity tests: to ensure that all analytical techniques used provide a correct declaration of analytes impurity content, such as related substances testing, heavy metals content, residual solvents content, and so on. Assay (content or potency): To provide a precise result, this enables accurate declaration of the content or potency of the analyte in a sample. For specificity validation, analytical techniques that can assess analyte response when all sample additives are present must be used. It is not always possible to prove that a single analytical technique is the only one that works for a certain analyte. In liquid chromatography, specificity is achieved by putting the chromatographic conditions, mobile section composition, column temperature, and detector wavelength along with the most effective columns. The pattern education step can be tuned for, in addition to chromatographic separation [10].
Limit of Detection (LOD): The smallest amount of analyte in a sample that can be detected but not necessarily quantified as an exact value is known as the detection limit. The lowest concentration level that can be identified statistically distinct from a blank at a certain degree of confidence is known as the limit of detection (LOD). The analysis of sample blanks is used to determine it. The smallest concentration of a substance that can be detected and reported with 99 percent confidence that the analyte concentration is greater than zero is known as the method detection limit (MDL). It’s calculated by analyzing a sample in a matrix that contains the analyte [10].
Limit of Quantification (LOQ): The lowest amount of analyte in a sample that can be quantitatively identified with sufficient precision and accuracy is known as the limit of quantitation (LOQ) or Quantitation limit of a particular analytical process [10].
Linearity & Range: The capacity of an analytical process to produce test results that are directly proportional to the concentration within a defined range is known as linearity. In the sample of analyte the range of the analytical technique should be used to analyze a linear connection. The proposed approach is used to show it directly on the drug substance by dilution of a standard stock solution of the drug product components. The confidence limit around the slope of the regression line is commonly used to express linearity. The ICH recommendation recommends a minimum of five concentrations to establish linearity.
Suitability: Machine suitability tests are a crucial component of chromatographic techniques, in line with the USP. These evaluations are used to confirm that the device’s resolution and reproducibility are sufficient for the evaluation to be carried out. The main premise of device compatibility tests is that the gadget, electronics, analytical activities, and samples make up a single unit that can be assessed as a whole. System performance is checked to ensure system performance prior to or during the evaluation of unknowns. This is known as system suitability. Determined parameters are compared to the requirements established for the technique, including plate dependence, tailing factors, resolution, and repeatability (%RSD retention duration and region of repetitive injection). These factors are assessed by looking at a machine suitability “pattern,” which may be a combination of primary additives and projected via-merchandise. The use of software created specifically for the purpose to offer a review of the separation and to provide a summary of the data on reproducibility may be used to carry out the documentation of device appropriateness. The software is also employed in the technique’s troubleshooting. To provide the feedback required to assess device performance, results saved in a relational database can be compared and summarized on a height-by-top or machine-by-device basis [13, 10].
Stability: Solution balance is the equilibrium of the well- known and sample answers that have been extracted from the sample or matrix and are ready to be injected, and it must be stored properly in both room temperature and refrigeration conditions depending on the stability of the pattern and well-known solutions. If the solution was refrigerated prior to investigation, it should have been thawed to room temperature before being tested for stability in both room temperature and the refrigerator. Two minimal examples of preferred and pattern responses need to be prepared, then they need to be examined using a unique methodology. The analyzed solutions are stored in the appropriate conditions, and depending on the nature of the product, stability may be established for a period of two days or on an hourly basis. Chemicals can break down before chromatographic examinations, for example, during the preparation of the sample solutions, extraction, cleanup, section transfer, or storage of organized vials (in refrigerators or in an automatic sampler). Below those circumstances, method development should evaluate the requirements’ and analytes’ stability. It is a measurement of the unfairness in the assay results produced at a certain point in a chosen period and language [14, 10].
Robustness: The robustness of an analytical procedure is a measure of its ability to remain unaffected by minor but deliberate modifications in method parameters, and it provides an indication of its dependability under normal conditions. If measurements are sensitive to changes in analytical circumstances, the conditions should be adequately controlled, or a precautionary remark should be included in the protocol. As a result of the robustness evaluation, a set of system suitability characteristics (e.g., resolution test) should be constructed to verify that the validity of the analytical technique is maintained whenever it is employed. Typical variables include: - analytical solution stability; - extraction time. In the case of liquid chromatography, typical variations include: - the effect of changes in pH in a mobile phase; - the effect of changes in mobile phase composition; - different columns (different batches and/or suppliers); - temperature; and - flow rate. Typical variations in gas chromatography include different columns (different lots and/or suppliers): - flow rate: - temperature [15, 16, 17, 18, 19].
Conclusion
This article discusses how to build a method, what validation is, the relevance of validation, the many types of validation, how to do the validation process, and its parameters to demonstrate that the method is suitable for its intended application. The basic goals of analytical technique development are identification, purification, and eventually qualification of any necessary medicine, etc. Analytical technique development aids in identifying crucial process factors and reducing their effects on precision and accuracy. Validation is a technique used in the pharmaceutical industry to verify that quality work is done in the process that supports the development of medicines and goods.
References
-
Piyush B, Ruchita B, Rushikesh B, Ganesh S, Kajal P et al. (2023) A review on specific and sensitive analytical method development and validation. OAJPR 7(4): 289.
-
Piyush B (2023) A review of high-performance liquid chromatography and its application. Journal of pharmacy and pharmaceutical science 12(13): 1-15.
-
Ravisankar P, Gowthami S, Rao GD (2014) A review on analytical method development. Indian J Res Pharm Biotech 2(3): 1183-1194.
-
Kissinger PT (2002) Instant Notes: Analytical Chemistry. Clin Chem 48(12): 2303.
-
Lloyd R, Snyder, Joseph JK, Joseph LG (1997) Practical HPLC Method Development 2nd (Edn.), John Wiley & Sons pp: 179-184.
-
McPolin O (2009) An introduction to HPLC for pharmaceutical analysis. Lulu.
-
Malviya R, Bansal V, Pal OP, Sharma PK (2010) High performance liquid chromatography: a short review. J Glob Pharma Technol 2(5): 22-26.
-
Jena AK (2011) HPLC: highly accessible instrument in pharmaceutical industry for effective method development. Pharm Anal Acta 3(1): 1000147.
-
Hamilton RJ, Sewell PA (1982) Introduction to high performance liquid chromatography. Springer pp: 1-12.
-
Priyanka S, Chatur, Usman (2021) Textbook of Pharmaceutical Validation M Pharm Sem-II S Vikas and Company.
-
Tranfo G, Enrico P, Renata S, Daniela P (2008) Validation of an HPLC/MS/MS method with isotopic dilution for quantitative determination of trans, trans-muconic acid in urine samples of workers exposed to low benzene concentrations. J Chromatogr B Analyt Technol Biomed Life Sci 67(1): 26-31.
-
Jenke DR (1998) Chromatographic method validation: A review of current practices and procedures. Part II. Guidelines for primary validation parameters. Instrum Sci Technol 26(1): 1-18.
-
Swartz E, Ira SK (1997) Analytical method development and validation. Marcel Dekker pp: 73-74.
-
Saranjit S, Monika B (2000) Guidance on Conduct of Stress Tests to Determine Inherent Stability of Drugs. Pharmaceutical Technology On-Line 24: 1-14.
-
Piyush b, Bachhav R, Sonawane D, Aher R, Deore R, et al (2023) The comparative study of different pharmacopoeias: Indian Pharmacopeia, British Pharmacopeia and United states Pharmacopeia. OAJPR 7(3): 285.
-
George L (2005) Methoxychlor, HPLC Methods for Recently Approved Pharmaceuticals 1st (Edn.), Wiley pp: 387-391.
-
Jay Br, Kelvin J, Pierre B (2003) Understanding and Implementing Efficient Analytical Methods Development and Validation. Bioprocess Online.
-
Chatwal GR, Anand SK (2002) Instrumental Methods of Chemical Analysis. Himalaya Publishing House.
-
Laxminarayan L, cswSharma N, Viswas A, Khinchi MP (2017) A review on chromatography techniques. Asian Journal of Pharmaceutical Research and Development 5(2): 1-8.
- Acido Labile or Gastro Irritant Apis and Enteric Release in Galenic Practice: An Overview
- A Study on Knowledge, Attitude and Practice of Hand Hygiene among Healthcare Professionals at a Tertiary Care Hospital, India
- Influence of Inoculum Concentration on In Vivo Incubation Period of Emmia lacerata, Pathogenesis and Management of Wilt in Pepper (Capsicum annuum L.)
- Vanilla’s Chemistry
- Marine Anti-Cancer Compounds and Adverse Effects of Global Warming on Oceans: An Overview
- Serological Investigation of Chikungunya Virus Antibody among Malaria-Suspected Febrile Patients in Some Healthcare Facilities in Rivers State