Revolutionizing FDA’s Quality Assessment of Regulatory Drug Applications with Knowledge-Aided Assessment and Structured Application (Kasa): An Innovative Approach
The regulatory evaluation process used by the FDA has changed through time, moving from a summary-based review in the 1990s to a question-based review and risk-based approach in the 2000s, and finally to an integrated quality assessment in 2015 and beyond. While attempting to ensure effectiveness, uniformity, and objectivity in its oversight of pharmaceutical quality, the FDA has faced difficulties. The FDA is developing a new approach called Knowledge-aided Assessment & Structured Application (KASA) to address these issues and make the most of technological advancements. The KASA system is intended to: 1) capture and manage knowledge throughout the lifecycle of a drug product; 2) develop rules and algorithms for risk assessment, control, and communication; 3) perform computer-aided analyses of applications to compare regulatory standards and quality risks across applications and facilities; and 4) provide a structured assessment that reduces textbased narratives and summarization of provided information. KASA is a new system designed to update the regulatory drug applications’ quality evaluation. KASA represents a conceptual change away from the out-dated evaluation procedures and toward a fresh, more effective method of managing data and resources. When completely developed and put into use, KASA will improve regulatory quality oversight’s efficacy, efficiency, and consistency through the lifecycle management of products and facilities and the sharing of information in a standardized and organized format. KASA will ultimately increase FDA’s emphasis on pharmaceutical quality, which is the cornerstone for guaranteeing the safety and efficacy of medications.
Introduction
The FDA’s Centre for Drug Evaluation and Research (CDER) is in charge of evaluating the effectiveness of medications that are sold to humans. All pharmaceuticals that are commercialized must be produced in accordance with defined quality criteria. Each dose must be free from contaminants and flaws and be both safe and effective. Patients and consumers can feel confident in their upcoming dose of medication because of its quality [1].
The Office of Pharmaceutical Quality (OPQ) was established by CDER in 2015 to: Establish a standardized procedure for ensuring quality across the whole drug product lifecycle (development → commercial manufacturing), Over the course of the drug’s life, evaluate all quality-related improvements that were suggested following application approval & Keep track of the quality of all regulated manufacturing facilities and pharmaceutical products [1].
An OPQ quality evaluation establishes if a drug’s formulation, production procedures, and manufacturing facilities provide a medication that is safe and effective for the target population. An OPQ quality assessment covers: Drug substance, Drug product and manufacturing: Process, Facilities, and Microbiology, Bio pharmaceutics [1].
From the summary-based review in the 1990s through the question-based review and risk-based approach in the 2000s to the integrated quality assessment in 2015 and beyond, the FDA has continued to develop its regulatory evaluation system. The challenge for the agency to effectively and reliably monitor pharmaceutical quality has grown as a result of the complexity of novel medication regimens and drug delivery systems. Practically speaking, due to initiatives from the 21 Century Cures Act, the FDA has seen a significant increase in the volume of regulatory drug applications with the reauthorization of the Prescription Drug User Fee Act (PDUFA VI), Bio similar User Fee Amendments (BsUFA II), and Generic Drug User Fee Amendments (GDUFA II). The FDA is looking to design a new system called Knowledge-Aided Assessment and Structured Application (KASA) in order to deal with these issues and manage the information influx [2].
The KASA system is a data-based platform for applications that assist knowledge management and structured quality evaluations. KASA is designed to: 1) Capture and manage knowledge during the lifecycle of a drug product; 2) Identify and communicate risks for the facilities and processes used to manufacture drug products using established rules and algorithms; 3) carry out computer-aided analyses of applications to compare regulatory requirements and quality concerns among the facilities and drug products in the repository of permitted items; 4) Offer a structured evaluation that completely obliterates narratives written in text and information summaries from applications [2].
The KASA system enables the FDA to record crucial assessment data as highly detailed structured data in a prescribed manner, enhancing the effectiveness, consistency, and objectivity of regulatory activities. KASA marks a substantial conceptual shift and revolutionizes the FDA’s capacity to implement sensible, thorough regulatory actions. In order to expand KASA to encompass drug substances, all generic dosage forms, new drug and biologic applications, and post-approval adjustments throughout the course of the next five years, the FDA will be seeking feedback at this advisory committee meeting. Additionally, the FDA will solicit feedback on the necessity of developing digitalization in KASA, including data standards and mobilization via cloud-based servers [3].
The Why of KASA
The agency is aware of the need for internal transformation to keep up with public demands, rising public expectations, and technical improvements in the twenty-first century. In the past, CDER assessments relied on freestyle narrative writing (word documents) that contained data that was copied and pasted, unstructured information, and a summary of the application. Such a framework hinders capacity to share knowledge and effectively manage the FDA’s array of approved drug items and facilities, and it may lead to inconsistency and inefficiency. Additionally, it limits ability to make decisions because assessors only consider a small portion of the vast amount of data available to the FDA when evaluating each application [2].
To address the aforementioned issues, OPQ created the KASA system to update the quality evaluation of drug applications and incorporate structured data. This encourages uniformity and makes it possible to use a crucial knowledge management tool, both of which enhance efficiency and the process of overall quality assessment [2].
![Figure 1: The KASA System [4].](/fulltextimages/11807/fig_1.png)
How is KASA designed to Work?
Initial Risk-Assessment
The OPQ staff will use KASA as a tool as part of their Integrated Quality Assessment (IQA) process to make quality-related regulatory decisions, such as approving drugs, creating responses to non-conformance situations, and maybe forecasting surveillance inspections. This tool depends on sponsors providing applications with pharmaceutical quality (PQ) and chemistry, manufacturing, and control (CMC) data and information in the appropriate parts of the eCTD. The entirety of eCTD Module 3 and the Quality Overall Summary (QOS) section of eCTD Module 2 will both contribute to KASA [5].
The FDA intends to have established standards for submitting PQ/CMC information within these application modules. When the agency receives the application, KASA will automatically be filled in with the data and details submitted in the appropriate submission modules. With the first risk-ranking calculation, the IQA procedure will begin. The critical quality attributes (CQAs) of the product and the initial inherent risk of that product will be taken into consideration when assessing this inherent drug product quality risk [5].
The intrinsic risk-ranking score for KASA will be determined by a computer-based algorithm and will take the following criteria into account:
- Information about drug substances and drug products
- Unit activities (related to CQAs)
- Scale-up
- risk to all quality areas (such as chemistry, microbiology, processes, and facilities)
- The firm’s or facility’s recognized skills (such information is anticipated to be summarized in the OPQ/Office of Quality Surveillance’s [OQS’s] facility dossier)
- Strict adherence to current good manufacturing procedures (CGMPs) (e.g., field alert reports, recalls, regulatory actions, etc.) and robustness of quality oversight
- Prior experience with unit operation or comparable items
- The results of previous inspections and the interval between inspections of the facility The initial risk will be ranked as low, medium, or high according to predetermined levels. For the purpose of calculating risk ranking in a quantitative risk assessment, failure modes, effects, and criticality analysis are proposed. It is suggested that the severity of the ranking be based on the chance of occurrence, the seriousness of the potential harm, and the possibility of being able to identify failure-related issues. In order to start doing their quality assessment, the regulatory assessor will get a structured, partially-populated (e.g., information auto-populated from the submission and/ or utilized to compute the risk-ranking score) IQA template from the risk-ranking component of KASA [5].
Risk Mitigation Assessment
The agency will conduct a quality evaluation of the drug product and its proposed manufacturing process using the KASA-generated, risk-ranked, applicable IQA template to make sure that all inherent hazards are as minimally affected as possible. This regulatory evaluation will be built to put a strong emphasis on risk reduction throughout the production process and from the facility’s perspective linked to the product’s CQAs. To avoid poor drug product quality or patient damage, assessment is focused on all areas with identified risks. Information obtained throughout the evaluation process and recorded in KASA is designed to be managed in terms of knowledge. Because KASA has the ability to manage knowledge during the assessment process, the more it is used and filled with data, the more knowledge the agency will have available regarding factors influencing their regulatory decisions and the more effective the tool will become at identifying risks associated with applications, products, or facilities [5].
As a result, the agency will be able to make regulatory choices based on product, facility, supply chain, or corporate risks to best utilize resources and guarantee that patients have access to high-quality, safe drug products [5].
Lifecycle Knowledge Management
As mentioned, one of the main goals of KASA is to provide FDA assessors with real-time data on the drug components, drug product, manufacturing processes, and facility information that will influence their assessment and regulatory decision-making, enabling them to conduct their reviews more effectively and efficiently [5].
The platform also aims to increase knowledge transparency across product lifecycle management, including better visibility into product, facility, and overall company quality. The evaluation team will have access to every phase of a product’s regulatory oversight, including the product’s initial approval in an NDA or BLA, all post-approval adjustments to supplements, and its transition to generic versions as ANDAs or biosimilars. Assessors will likely be equipped with facility dossiers in addition to KASA to provide additional high- quality information. When these solutions are used together, knowledge management between desk application assessors and what investigators see during facility inspections should be greatly enhanced [5].
Information is currently kept in a number of repositories for product quality evaluation, which is carried out by CDER, and facility inspection, which is overseen by the Office of Regulatory Affairs for small molecule products. Not all relevant staff members always have easy access to information. Since the inspection data will be fed directly into the program, KASA should make it easier for desk assessors to retrieve inspection data. Real-time facility dossiers are being created by OQS using all available data on drug products, facilities, and corporate quality, which is also pertinent to KASA and IQA. Control scheme outliers can be found by processing this information through a potent informatics system [5].
KASA and associated facility dossiers will probably have an impact on other regulatory operations, including carrying out pre-approval inspections, completing product- specific post-approvals, or planning surveillance inspections. KASA will let assessors know about any inherent hazards in a product or current issues with a facility or business (for example, in the most extreme situations, a problem with data integrity). In order to guarantee that the inspection concentrates on high-risk areas, this will enable inspection suggestions to be made and any risks found to be conveyed to the investigator [5].
In contrast, desk assessors will have quick access to inspection information to confirm that risk mitigation has been proven at the plant or warn assessors of problematic areas that weren’t immediately evident in the initial risk assessment or the application. To make sure agency resources are being utilized to their full potential for pre- approval and surveillance inspection planning, information from KASA and/or facility dossiers can also be used. This will guarantee that inspection resources are concentrated on the sites, production processes, and products that pose the most risk [5].
![Figure 2: KASA’s connection with other relevant OPQ’s Initiatives/Programs [4]. (QSD: Quality Surveillance Dashboard, IQA: Integrated Quality Assessment, ICH M4Q (R2): The Common Technical Document – Quality, PQ/CMC: Pharmaceutical Quality/Chemistry Manufacturing and Controls).](/fulltextimages/11807/fig_2.png)
Structured Application
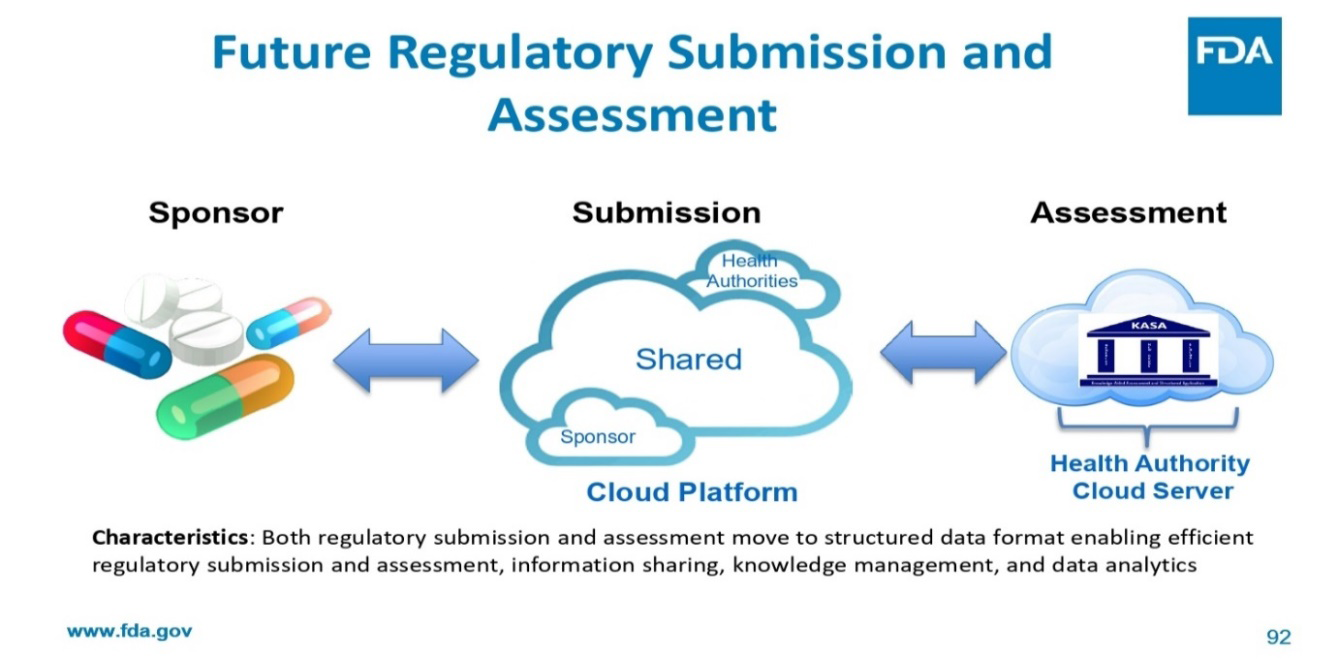
In the future, applications submitted with streamlined layouts and organized data that link with the assessment system would considerably improve knowledge-aided evaluation. The FDA presently accepts regulatory drug applications in the electronic common technical document (eCTD) format. Because the submitted content does not adhere to the development flow, contains unstructured data, and varies in the amount of granularity offered, the eCTD causes difficulties for FDA assessors despite its great advantages. Additionally, because the documents are in PDF format, it is difficult to search for and mine the information, which makes lifecycle management difficult [2].
The modification of ICH M4Q (R1) and pharmaceutical quality electronic data standards are two ongoing projects that will help with the “structured application” portion of KASA. It is possible that recommendations for submission structure will be provided in the future to better integrate with KASA’s structured evaluation process. Applicants would then be able to concisely and consistently list the efforts taken to reduce inherent hazards through development studies and control methods. According to this paradigm, the KASA template would be filled out automatically using tools to extract information from the structured submission, such as specifications and essential process parameter ranges. By freeing the assessor from administrative duties, this would increase assessment effectiveness by enabling assessors to concentrate on high-risk areas [2].
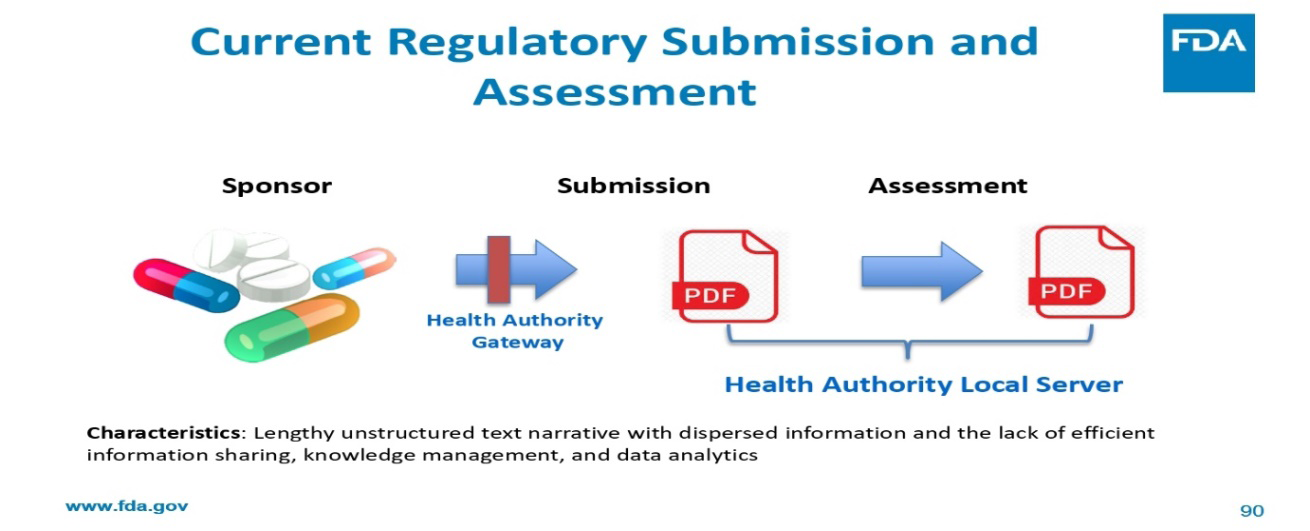
- Submission and Assessment
- Assessment is moving into cloud
- Submission and Assessment
- Lengthy unstructured text narrative with dispersed information and the lack of efficient information sharing, knowledge management, and data analytics
- Regulatory assessment moves to structured data enabling efficient information sharing, knowledge management, and data analytics, resulting efficient regulatory assessment
Table 1: Comparison of Characteristics.
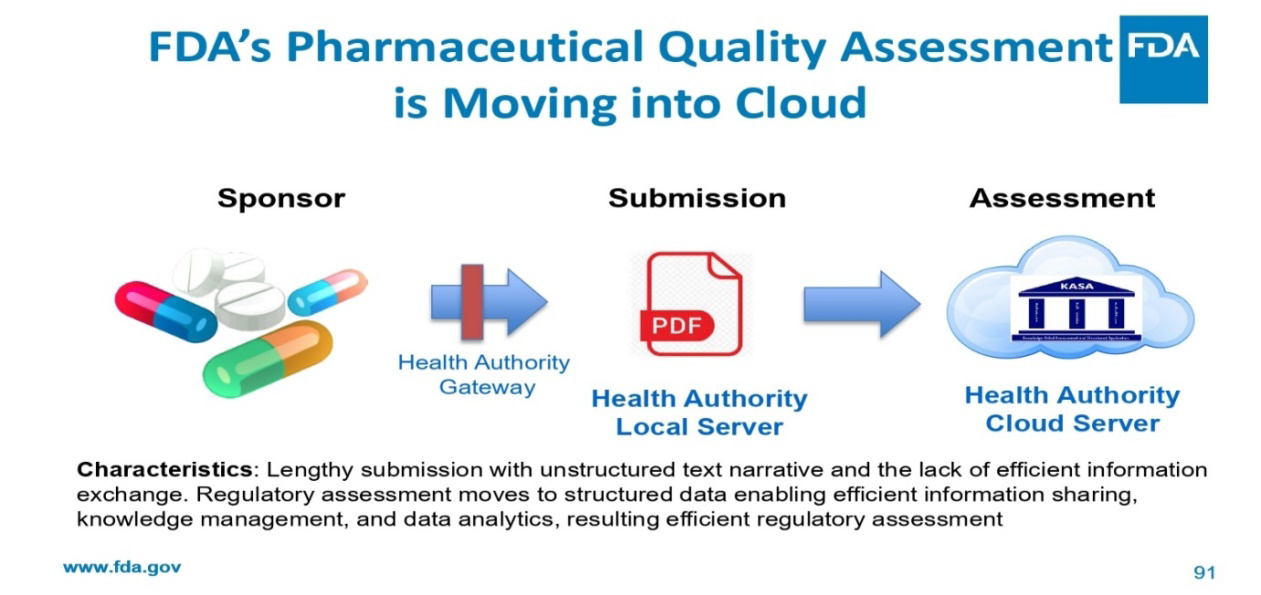
- Submission and Assessment
- Assessment is moving into cloud
- Submission and Assessment
- Lengthy unstructured text narrative with dispersed information and the lack of efficient information sharing, knowledge management, and data analytics
- Regulatory assessment moves to structured data enabling efficient information sharing, knowledge management, and data analytics, resulting efficient regulatory assessment
Table 1: Comparison of Characteristics.
Benefits Offered by KASA
The KASA system replaces the present unstructured text document used for regulatory application assessment with an issue-based regulatory and technical assessment that uses structured data and information with standardized formatting, common terminology, and a uniform output.
Both regulatory submission and assessment move to structured data format enabling efficient regulatory submission and assessment, information sharing, knowledge management, and data analytics This enhances knowledge management inside the agency as well as the consistency, transparency, communication, and objectivity of regulatory activities [2].
With access to structured knowledge, KASA provides technologies that allow assessors to automatically extract historical data and facility information to better inform the regulatory review and decision-making process. By utilizing rules and algorithms, KASA makes it easier to assess risk while lowering the amount of effort and subjectivity required for documentation. The built-in rules and algorithms, along with the outlier detection, enable assessors to concentrate on high-risk issues and areas, which enhances the effectiveness and quality of the regulatory evaluation. Finally, KASA conveys the remaining product, manufacturing, and facility risk for each regulatory submission by assessing hazards and mitigation measures. The agency’s evaluation of post- approval adjustments and the lifecycle management of drug products are made easier with the agency’s assistance in clearly defining the key mitigating factors and residual risk. This can assist in concentrating resources for post-approval and follow-up inspections on the riskiest products [2].
Where KASA is today
By utilizing 1) structured data (as opposed to narrative information), 2) sophisticated analytics, and 3) knowledge management, OPQ’s KASA system was envisioned as a way to modernize FDA evaluation in 2016. Multiple OPQ and CDER co-workers worked diligently and in partnership to develop this KASA concept throughout multiple revisions. Through the development, testing, and application of KASA prototypes over time, the KASA vision has been improved [2].
The FDA moved KASA to the cloud in 2020, storing KASA quality evaluations on FDA servers in a FISMA-high environment with the highest level of security to guarantee the preservation of sensitive data. The FDA is utilizing the adaptability and speed of cloud computing by shifting the quality evaluations there, allowing for effective knowledge management and data analytics using structured data. This big KASA launch, known as KASA 3.0, represents an important milestone in the modernization of quality assessment as a whole. KASA 3.0, which is used to evaluate the quality of generic solid oral dosage forms, marks an important development and implementation milestone. In order to operationalize KASA, which is now used by three assessment disciplines (drug product, manufacturing, and bio pharmaceutics) for the quality evaluation of generics, OPQ has made great strides [2].
On continuing KASA’s development, OPQ is concentrated. Following the launch of KASA 3.0 for generics, long-term goals include extending KASA to drug substances, all generic dosage forms, novel medications, biologics, and post- approval adjustments throughout the course of the following five years [6].
Conclusion
KASA is a system created to update the way regulatory drug submissions are evaluated for quality. KASA symbolizes a paradigm shift away from the antiquated assessment procedures of the past and toward a new, more effective method of managing data and resources. Through knowledge management, KASA helps to:
- Ensure patient-focused quality standards and the objectivity of regulatory actions.
- Improve science- and risk-based regulatory approaches and
- Improve regulatory oversight through lifecycle management of products and facilities.
The KASA method ultimately enhances OPQ’s emphasis on pharmaceutical quality, the cornerstone for guaranteeing the safety and efficacy of medications. Through modernization, it elevates the agency’s quality control to the next level [7].
References
-
Yu L (2019) Pharmaceutical Product Development: Evolving Regulatory Landscape. 4th PQRI/FDA Conference on Advancing Product Quality, Bethesda, MD, USA, pp: 1-36.
-
(2023) FDA Briefing Document, Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting, pp: 1-16.
-
Chatterjee B (2019) Understanding the FDA’s Knowledge-Aided Assessment Structured Application (KASA) Framework.
-
Lee SL (2022) Knowledge-aided Assessment & Structured Application (KASA): A New Approach that Modernizes FDA’s Quality Assessment of Regulatory Drug Applications, pp: 1-108.
-
Demystifying FDA’s KASA Initiative… and how it aims to improve drug product, facility, and corporate quality monitoring.
-
Woodcock J (2018) Efficient Generic Drug and Biosimilar Review and Surveillance Processes, Association for Accessible Medicines, pp: 1-26.
-
Raw A (2022) The Future of FDA Quality Assessment Knowledge-Aided Assessment & Structured Application – KASA. DMF Workshop: GDUFA III Enhancements and Structured Submissions, pp: 1-36.
- Hydrogen Peroxide Scavenging by Methanolic Extracts of Coriander: An In Vitro Antioxidant Study
- Aromatherapy in Palliative Care: A Fragrant Quest for Relief
- Empowering Women, Securing Futures: Contraception’s Role in Socioeconomic Progress in India
- Effect of Crospovidone, Croscaramellose Sodium in Combination on the Drug Release of Anti diabetic Medication in Tablet Form
- Knowledge, Attitudes, Anxiety, and Preventive Behaviors Regarding Covid-19 Affliction among Healthcare Workers in Pakistan
- “Competitive Landscape and Brand Equivalents: Implications for ANDA (Abbreviated New Drug Application) Approval”