We are Reporting an Infant with Genito Patellar Syndrome and KAT6B Gene Mutation Presenting with Facial Dysmorphism, Bilateral Genu Recurvatum, Hip Dislocations, Absence of Cavum Septum Pellucidum, Anal Abnormality and Hydronephrosis
A preterm female infant was admitted to neonatal intensive care unit with respiratory distress, facial dysmorphism, bilateral genu recurvatum, hip dislocations, absence of cavum septum pellucidum, anal abnormality, and hydronephrosis. Exome study confirmed the diagnosis of Genito patellar syndrome.
Introduction
Genito patellar syndrome is a rare condition seen in 1 in a million new-born infants and is typically characterized by genital abnormalities, absence or abnormality of patellae, developmental and intellectual delay and many other associated abnormalities [1]. KAT6B gene mutation is associated with this disorder [2]. Typically, males present with cryptorchidism and poorly developed scrotum and females may present with clitoromegaly and hypoplasia of labia. We are reporting an infant with Genito patellar syndrome with KAT6B gene mutation presenting with facial dysmorphism, bilateral genu recurvatum, hip dislocations, absence of cavum septum pellucidum, anal abnormality, failed hearing screen and hydronephrosis.
Our patient presented as a newborn female infant delivered at a gestational age of 30 weeks and 3 days to a 37-year-old, gravida 6 and para 4 mother via precipitous vaginal delivery. APGARS scores were 3 at 1 and 9 at 5 minutes. Prenatal studies were suggestive of limb length discrepancy, absence of cavum septum pellucidum, ventriculomegaly and intrauterine growth retardation. Pregnancy was complicated by chronic hypertension, gestational diabetes, maternal hepatitis-C antibody positive and maternal urine screen positive for marijuana metabolites. The infant was noted to have respiratory distress soon after birth, intubated and transferred to the NICU. Birth weight was 1260 grams, length was 45cm, and head circumference was 32cm. The infant was weaned off the respirator and switched to bubble CPAP on day 1 of life. She did not require surfactant administration. She continued to require non-invasive ventilation. On follow- up, she had clinical and radiological evidence of chronic lung disease. She was intubated and was on respirator for several days following anaesthesia for closed reduction of bilateral hip dislocations, neuro imaging, and gastrostomy. She was initially on intravenous furosemide, as needed. She was switched to oral diuretics, spironolactone and chlorothiazide at 4 months of age. Over the next few weeks, FiO2 was gradually weaned to low flow nasal canula. She continued to require supplement oxygen until discharge. She was discharged home on oral diuretics.
Renal sonogram was consistent with grade 3 bilateral hydronephrosis. She had an episode of E Coli urinary tract infection. Following this episode of urinary tract infection, she was on prophylactic amoxicillin. However, while on prophylactic amoxicillin, the infant had another bout of E coli urinary tract infection. Voiding cystourethrogram was negative for vesicoureteral reflux. She was discharged home on sulfamethoxazole and trimethoprim for urinary tract infection prophylaxis. She will have a follow-up by a pediatric urologist after discharge from the hospital.
Prenatal studies were suggestive of absence of cavum septum pellucidum and mild ventriculomegaly. Head ultrasound confirmed dysgenesis of the corpus callosum with normal looking ventricles. Brain MRI confirmed complete agenesis of the corpus collosum and possibly polymicrogyria. She had a sacral midline dimple. Spinal ultrasound indicated borderline low position of the conus medullar at mid lumbar 3. MRI of the spine showed foreshortening of the posterior C1 arch resulting in moderate foramen magnum stenosis. Trace fluid was noted posterior to the ligamentum flavum, likely interposed between the ligament and the C2 posterior elements that was thought to be a congenital or acquired stripping from the adjacent osseous structures. Craniovertebral junction and upper cervical ligaments otherwise appeared intact. The neurosurgical team had minimal concerns for C1/C2 instability. Thyroid studies, ACTH/Cortisol, and IGF1 and IGF3, estradiol, and LH/ FSH all were within normal limits around 3 months of age. Endocrinology follow-up was recommended at 6 months of age due to risk of thyroid and pituitary disorders associated with GPS and agenesis of corpus callosum.
Nipple feeds were attempted on several occasions but were unsuccessful. She could not move her neck into flexion and maintained it in a relatively extended position. There was a concern for C1/C2 instability and possible cervical spine subluxation. CT C-spine was performed confirmed hyperextension Figure A.
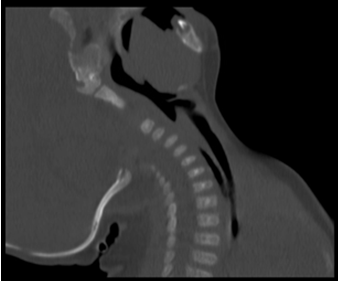
Figure A: CT Spine Demonstrating Hyperextension of C1/C2 while in Neutral Position.
She was gavage fed for several months. Swallow studies were negative for aspiration of feeds. She had a gastrostomy and at the time of discharge she was on 26 calorie Elecare feeds. She was on bolus feeds from 6AM to 8PM and on continuous feeds at night with a goal of total fluids at 150ml per Kg.
Initially, she presented with genu recurvatum (Figure B) with dislocated hips bilaterally, rotation of the ankles, anterior anus Figure C.
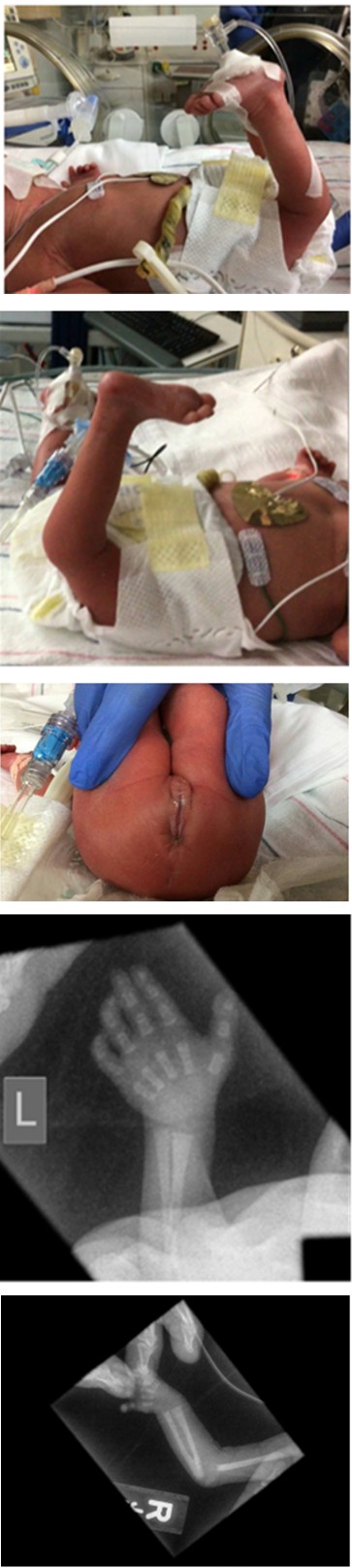
Figure B: Gene recurvatum Noted on DOL 1.
Figure C: Anterior Anus.
Single transverse palmar crease and high arched palate. The skeletal survey demonstrated radial ulnar synostosis (Figure D-Right), bilateral elbow subluxation, and foreshortened metacarpals bilaterally Figure D-Left.
Figure D: radial ulnar synostosis and foreshortened metacarpals Joints on Skeletal Survey.
Although genu recurvatum was noted clinically, the knees appeared grossly unremarkable. Physical therapy showed mild improvement in hip and elbow motion. There was little improvement in the range of motion at both knees. Ultrasound revealed both knees were reduced but hyperextended and she was noted to have bilateral hip dislocations Figures E and F.
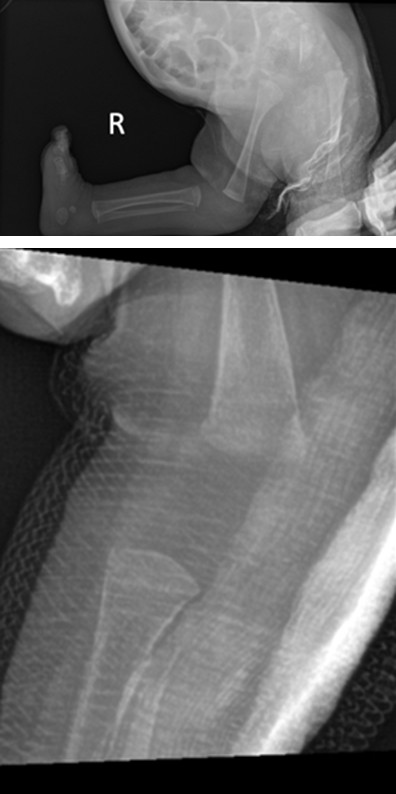
Figure E: X-ray Demonstrating hyperextension at the Right Knee.
Figure F: Post-Closed Reduction X-ray of R Leg Demonstrating Improvement in Hyperextension of the Knee.
Pavlik harnesses were attempted without much success. A surgical closed reduction was done with bilateral splints. Splints were causing pressure sores on heels with minimal benefit to her range of motion. Orthopedic surgeon recommended follow up at 6 months for open reduction.
She had a soft systolic murmur at the time of admission. Initial echocardiogram showed moderate patent ductus arteriosus of 3mm with left to right shunting, dilated right atrium, and tricuspid regurgitation. A follow-up study showed patent ductus arteriosus measuring 2 mm, with left to right shunting, and bilateral branch pulmonic stenosis.
Genetic study on exome sequencing was positive for KAT6B gene mutation. The initial newborn screen showed elevated IRT without CFTR mutation. Repeat studies had persistently elevated IRT. After exome sequencing, she was diagnosed as an unaffected carrier of CFTR mutation. She will be followed up by a pediatric pulmonologist and a sweat chloride test is being arranged.
Discussion
Our index patient presented with skeletal dysplasia, bilateral genu recurvatum, hip dysplasia and knee dislocations, facial dysmorphism. The differential diagnosis included Genito patellar syndrome, Say Barber Biesecker Young Syndrome, nail patella syndrome, Mowat Wilson syndrome and Larsen syndrome.
Genito patellar syndrome and Say Barber Biesecker Young Syndrome are associated with the KAT6B gene mutation [3, 4]. This gene is important for synthesis of histone acetyltransferase. This enzyme modifies histones, important structural proteins that attach to DNA and give chromosomes their shape. KAT6B appears to regulate genes that are important for early development, including development of the skeleton and nervous system. Both disorders present with marked developmental deficits, mental retardation, patellar hypoplasia or aplasia. There is an association of diffuse hypotonia and varying degrees of genital abnormalities. Skeletal defects are joint dislocations at hips and knees; flexion contractures of the knees and hips, or genu recurvatum. There may be associated chest anomalies, digital hypoplasia and spine abnormalities. All reported cases of GPS have resulted from new mutations in the gene and have occurred in people with no history of the disorder in their family. In SBBYS syndrome KAT6B mutations cluster in a ∼1,700 base pair region in the 3′ part of the large exon 18. GPS has KAT6B mutations located in the 5′ region of the same exon [5]. Penetrance appears to be complete. GBS and SBBYS disorders related to KAT6B require molecular genetic testing. KAT6B disorders are inherited in an autosomal dominant manner. To date, most individuals with a KAT6B disorder have had a de novo pathogenic variant. Exome sequencing is the most used method to establish the genetic defect [6]. Prenatal and preimplantation genetic testing are possible for families in which the pathogenic variant has been identified. Congenital heart defects, small bowel malrotation, feeding difficulties, slow growth, cleft palate, hearing loss, and dental anomalies have been observed in individuals with either phenotype.
SBBYS typically presents with vertical narrowing and shortening of the palpebral fissures, ptosis, intellectual disability, hypothyroidism, hearing impairment, and dental anomalies. SBBYS phenotypes may have long thumbs and long great toes and lacrimal duct abnormalities. There are multiple skeletal abnormalities like GPS. Both GPS and SBBYS disorders have global developmental deficit. Treatment focuses on the associated skeletal, cardiac, spine or rib cage abnormalities. Our index patient required prolonged mechanical non-invasive respiratory support. Bilateral genu recurvatum with associated knee and hip dislocations provided varying degrees of challenges.
Closed reduction of hip dislocation was of limited help for our patient and additional open reduction surgery was scheduled. Early physical therapy is indicated for joint contractures and dislocations, talipes equinovarus deformities may necessitate splinting and stretching. It is recommended to advocate early speech therapy and physical therapy to increase range of motion of joints followed by enrollment in early childhood intervention and special education programs for developmental delays and intellectual disability. Both GPS and SBBYS may have hypothyroidism or gastrointestinal problems like malrotation of bowel, megacolon or Hirsch Sprung disease. Common features of both are arthrogryposis multiplex congenita, clitoral hypertrophy, coarse facial rounded and heavy features or thickened skin with or without thickening of subcutaneous and bony tissues, and cryptorchidism [7].
GPS has higher incidence of genital anomalies, anal anomalies, flexion contractures at hips and knees, agenesis of corpus callosum with microcephaly. On the other hand, SBBYS has long thumbs, mask like face, lacrimal duct anomalies, hydronephrosis or multiple renal cysts. Both disorders have patellar hypoplasia/aplasia, Congenital cardiac defect, developmental delay, dental anomalies, hearing deficit, hypothyroidism, and hypotonia [8]. Currently, the oldest known individual diagnosed with KAT6B is in her 40’s. The motor and intellectual deficit is usually severe; some can barely communicate through vocalization, few patients may be able to manipulate objects, and ambulate with walkers or tricycles. Behavioral problems may be autism spectrum disorder, anxiety, aggressive behavior, and attention deficit. Most patients have severe microcephaly, hydronephrosis and cystic kidneys. Prenatally, they may have oligohydramnios sequence [9]. Abnormal palmar creases, widely spaced nipples, hypoplastic nails, and café au lait macules have been reported in a few affected individuals. Bashir et al, reported sagittal craniosynostosis in two individuals with the intermediate phenotype [10].
Meier-Gorlin syndrome is an autosomal recessive disorder that presents with patellar aplasia or hypoplasia; microcephaly; genital anomalies; contractures, severe intrauterine & postnatal growth restriction and bilateral microtia. Additional features are a small mouth, micrognathia, full lips, and a narrow nose with a high nasal bridge. Meier- Gorlin syndrome can be caused by mutations in one of several genes: ORC1, ORC4, ORC6, CDT1, and CDC6. Conductive hearing loss has been reported in some individuals with narrow ear canals. Growth delay may benefit with growth hormone administration. Despite a small head size, most people with Meier Gorlin syndrome have normal intellect [11].
Nail-patella syndrome presents with changes in the nails, knees, and elbows, and the presence of iliac horns [12]. The disorder is related to variants in the LMX1B gene. Nails may be poorly developed, abnormal or absent. The patellae may be small, irregularly shaped, or absent. There may be elbow abnormalities associated with limited movements and cubitus valgus. Iliac horns have been reported, they are bilateral, conical, bony processes that project posteriorly and laterally from the central part of the iliac bones of the pelvis. The disorder may be associated with renal abnormalities, glaucoma and ocular hypertension.
Mowat-Wilson syndrome (MWS) is an autosomal dominant disorder caused by a pathogenic variant in ZEB2, a heterozygous deletion of 2q22.3 involving ZEB2 [13]. This disorder is manifested by facial dysmorphic features of hypertelorism, thick eyebrows, pointed chin, open-mouth, and abnormal ears. There may be associated congenital cardiac defects to include abnormalities of pulmonary valves. Hirschsprung disease or chronic constipation, genitourinary anomalies, abnormalities of corpus callosum. There is a higher incidence of superior developmental delay, and speech delay with limited to a few words. Mostly receptive language skills are established. Growth delay with microcephaly and seizure disorder are reported. Facial dysmorphism with Hirschsprung disease and or chronic constipation, developmental delay, and intellectual disability can establish the clinical diagnosis. Further genetic studies are confirmatory.
Larsen syndrome is a disorder of the skeletal system that typically presents with dislocations of hips, knees, elbows. Talipes equinovarus deformity, contracture or hypermotility of joint has been reported. There is a mutation or deletion in FLNB gene which interferes with the proliferation or differentiation of chondrocytes. This disorder is not associated with absence of corpus callosum [14]. Tips of the fingers and thumb may be square-shaped. Facial dysmorphism includes frontal bossing flat bridge of the nose and midfacial hypoplasia with hypertelorism. Cardiac and renal anomalies may be associated. Most have normal intellect. The disorder is inherited as autosomal dominant pattern. De Novo new mutations have been reported.
Conclusion
Multi system involvement with GPS requires careful attention for prolonged respiratory support, management of skeletal defects, eye examination, endocrine work up, renal assessment and cardiology assessment. Exome study is the diagnostic tool to confirm the diagnosis. Our index infant had significant respiratory problems requiring careful management during hospital stay followed by a discharge on oral diuretics and supplemental oxygen. Referral was made to pediatric pulmonologist to manage chronic lung disease, follow-up for any adjustments in diuretic needs management of electrolytes and potential management of risk of osteopenia.
Hypothyroidism is a potential risk factor and follow- up thyroid functions is required. We referred our patient to an endocrinologist for any thyroid or pituitary related disorders. Basic endocrine workup during hospital stay was unremarkable. We made a referral to an early intervention program to access occupational, physical, speech, and feeding therapy. Orthopedic intervention with open reduction of hip dislocation is planned at the age of 6 months. The Infant will require follow-up by Shriners children hospital. Physical therapy will be continued after discharge to increase joint mobility. Our infant failed auditory brain evoked response x3. An arrangement has been made to get follow-up by an audiologist for a detailed workup and support. We will follow developmental progress and educational needs; nutritionist is going to follow feeding issues. The initial eye exam and retinal assessment was normal, we have arranged a follow- up with the pediatric ophthalmologist.
References
-
Medline Plus (2013) Genitopatellar syndrome.
-
Campeau PM, Lu JT, Dawson BC, Fokkema IF, Robertson SP, et al. (2012) The KAT6B related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum Mutat 33(11): 1520-1525.
-
Medline Plus (2013) Genetics.
-
Medline Plus (2013) Ohdo syndrome, Say Barber Biesecker Young Simpson variant.
-
Szakszon K, Salpietro C, Kakar N, Knegt AC, Olah E, et al. (2013) De novo mutations of the gene encoding the histone acetyltransferase KAT6B in two patients with Say-Barber Biesecker Young-Simpson syndrome. Am J Med Genet 161A(4): 884-888.
-
Lemire G, Campeau PM, Lee BH (2012) KAT6B Disorders. GeneReviews [Internet] Seattle (WA): University of Washington, Seattle 1993-2024.
-
National Center for Advancing Translational Sciences (2024) Genitopatellar syndrome.
-
Okano S, Miyamoto A, Fukuda I, Tanaka H, Hata K, et al. (2018) Genito patellar syndrome: the first reported case in Japan. Hum Genome Var 5: 8.
-
Kim BR, Han JH, Shin JE, Park MS, Park KI, et al. (2019) Genito patellar syndrome secondary to de novo KAT6B mutation the first genetically confirmed case in South Korea. Yonsei Med J 60(4): 395-398.
-
Bashir RA, Dixit A, Goedhart C, Parboosingh JS, Innes AM (2017) Lin Gettig syndrome Craniosynostosis expands the spectrum of the KAT6B related disorders. Am J Med Genet 173(10): 2596-2604.
-
de Munnik SA, Bicknell LS, Aftimos S, Al-Aama JY, van Bever Y, et al. (2012) Meier Gorlin syndrome genotype phenotype studies 35 individuals with pre replication complex gene mutations and 10 without molecular diagnosis. Eur J Hum Genet 20(6): 598-606.
-
Sweeney E, Hoover Fong JE, McIntosh I (2003) Nail Patella Syndrome. Gene Reviews.
-
National Organization for Rare Disorders (2020) Mowat Wilson Syndrome.
-
Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, et al. (2007) Guidelines for the diagnosis and management of individuals with neurofibromatosis. J Med Genet 44(2): 81-88.
- Understanding Pediatric Multiple Sclerosis: Clinical Presentation, Diagnostic Criteria, Therapeutic Advances, and Supportive Care Approaches
- Hemophilia in Children
- Xia-Gibbs Syndrome- A Case Report
- A Study to Assess Effectiveness of Play Therapy in Reducing Post-Operative Pain among Children Age 2 To 5 Year who have Undergone General Surgeries in Selected Pediatric Hospitals of Vadodara
- Preterm Birth: Scope of the Problem, Cost of Care, Potential Complications and Current Guidelines for Management
- Noradrenaline: Can we Use it to Manage Hemodynamic Instability among Neonatal Septic Shock at the NICU?