Zimmerman Laband Syndrome and its Spectrum Disorders: Mini Review
Combination of hypertrichosis, gum hypertrophy, hypoplasia/ aplasia of nails and coarse facial features are common features of group of genetic disorders. Subtle clinical feature variation may be useful for differentiating all. This review article will be useful for the clinical diagnosis of these conditions.
Abbreviations
ZLS: Zimmerman Laband Syndrome; TBS: Temple-Baraitser Syndrome; CdLS: Cornelia de Lange Syndrome; NPS: Nail Patella Syndrome; CSS: Coffin-Siris Syndrome.
Introduction
There are few rare genetic syndromes characterized by similar set of presentation like hypertrichosis, gum hypertrophy, hypoplasia/ aplasia of nails and coarse facial features. However there remains subtle difference among each one of them that helps to differentiate one from another clinical entity. There are many syndromes which share mutation (KCN1) in similar genes or may arise de novo. We labelled them as Zimmerman Laband Syndrome and associated spectrum disorders. These syndromes are Zimmerman Laband Syndrome (ZLS), Temple-Baraitser syndrome (TBS), Wiedemann Steiner Syndrome, Cornelia de Lange syndrome (CdLS), Nail Patella Syndrome (NPS) and Coffin-Siris syndrome (CSS). Here we present Zimmerman Laband Syndrome and associated spectrum disorders. They have subtle difference among each one of them that helps to differentiate one from another clinical entity. Here we describe 6 related clinical syndromes.
Zimmerman Laband Syndrome (ZLS)
Zimmerman Laband Syndrome is a rare autosomal dominant congenital disorder. It is characterized by gingival hypertrophy, hypo/aplastic nails and distal phalanges, hypertrichosis, abnormalities of soft cartilages of the nose and/or ears, hepatosplenomegaly, hyperextensibility of joints, mild hirsutism, hypertrichosis and intellectual disability [1]. It affects both sexes equally. Typically, gingival fibromatosis is present at birth or manifests soon after. The maxillary and mandibular gingiva grow gradually over time. Genetic variation exists in ZLS. There have been reports of heterozygous gain of function missense variants in the KCNH1 (1q32.2) and KCNN3 (1q21.3) genes as well as, less frequently, recurrent missense variants in the ATP6V1B2 (8p21.3) gene [2, 3]. It is very difficult to diagnose ZLS in neonatal age group as many symptoms may not present very early like intellectual disability, convulsions, gingival hypertrophy. As the child grows, these problems start to manifest. A detailed medical history and physical are characteristic of this syndrome. Other differentials include cross syndrome with hypopigmentation, oligophrenia, and athetosis, Rutherford syndrome, Cowden syndrome and Murray syndrome. Antenatal exposure of inflammation, pregnancy, leukaemia, or drugs such as phenytoin, diltiazem, cyclosporine A, verapamil and nifedipine must be ruled out for acquired causes. Mostly patient presents at later age in surgical/ dental OPD due to problems arising because of gingival hypertrophy, cosmetic concerns, difficulty in eating, etc (Figure 1).
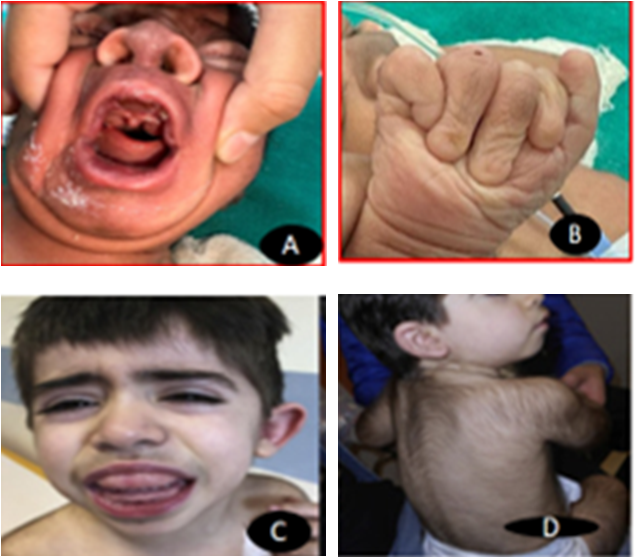
Figure 1A: Board nasal bridge and wide philtrum 1B: Absent nails in right hand coarse facies 1C: wide philtrum 1D: Hypertricosis.
Temple-Baraitser Syndrome (TBS)
TBS and ZLS are rare developmental disorders [4]. TBS is characterized by clinical features being intellectual disability, gingival fibromatosis, hypoplasia of the distal phalanges, hirsutism, scoliosis, hepato-splenomegaly, and absence or dysplasia of all nails. In contrast, ZLS has a pseudo-myopathic look, hypotonia in infancy, hypoplasia/aplasia of the thumb and great toe nails and epilepsy.
Wiedemann Steiner Syndrome
It is a rare autosomal dominant disorder with a variable clinical phenotype and de novo pathogenic variations in the KMT2A gene. The syndrome appears to be prevalent in all populations and affects both sexes equally [5]. Hypertelorism, a wide nasal bridge, a long philtrum, thick eyebrows, synophrys, and long eyelashes are typical facial traits. Patients present with mild to moderate intellectual disability, developmental delay, behavioural problems, and hypertrichosis cubiti or generalized hypertrichosis. Sacral dimple, rib abnormalities, and vertebral block have been reported in some patients.
Cornelia de Lange Syndrome (CdLS)
It can be inherited either an X-linked or autosomal dominant. Seven genes the NIPBL gene on chromosome 5, the SMC1A gene on the X chromosome, the SMC3 gene on chromosome 10, the Rad 21 gene on chromosome 8, the HDAC8 gene on X chromosome, the ANKRD11 on chromosome 16, and the BRD4 gene on chromosome 19 have been linked to CdLS [6]. The manifestations are synophrys, long eyelashes, limb deformities, growth retardation, intellectual impairment, and generalized hirsutism [7]. Numerous additional symptoms are also present with the condition, including hearing loss, myopia, cardiac septal abnormalities, gastrointestinal disorders, autism and self- destructive conduct, and cryptorchidism or hypoplastic genitalia. Treatment requires a multidisciplinary approach including gastroenterologist, neurologist, otologist, nutritionist, endocrinologist, cardiologist and involvement of other domains such as physical therapy, speech therapy, or occupational therapy to improve development and long- term prognosis.
Nail Patella Syndrome (NPS)
It is an autosomal dominant disorder which occurs due to a heterozygous loss of function mutation of the LMXB1 gene on chromosome 9q34.1 A multi-systemic disease characterized by clinical tetrad of elbow abnormalities, presence of iliac horn, hypoplasia or absence of the patella, and dysplasia of the fingernails. However ocular involvement in the form of glaucoma, renal involvement in the form of nephropathy, and neurological involvement in the form of neuropathic pain and numbness may exists as well [8]. Progression of proteinuria may be blocked by renin angiotensin aldosterone system and surgery may be required for patellar instability.
Coffin-Siris Syndrome (CSS)
The pathogenic gene variants are ARID1A, ARID1B, ARID2, SMARCA4, SMARCB1, SMARCE1, SMARCC2, DPF2, SOX4 and SOX11 [9]. CSS has fifth-digit nail/ distal phalanx hypoplasia/aplasia [10]. developmental or cognitive delay, coarse facial features demonstrating wide mouth with thick, everted upper and lower lips, broad nasal bridge with broad nasal tip, thick eyebrows, and long eyelashes, central hypotonia, hirsutism/hypertrichosis and sparse scalp hair. Occupational, physical, and/or speech therapies can be used to treat symptoms in order to maximize developmental results. To address nutritional demands, feeding treatment, nutritional supplements, and/or the implantation of a gastrostomy tube may be necessary. Regular follow up and management of hearing loss and ophthalmologic problems is required.
Facial Dysmorphism, Hypertrichosis, Epilepsy, Intellectual/developmental delay, and Gingival overgrowth (FHEIG) Syndrome. It is an autosomal dominant condition characterized by variable intellectual disability, epilepsy, global hypertrichosis, severe gingival overgrowth, vision impairment and congenital anomalies/dysmorphic syndrome. The gene implicated for this disease is the mutation in Potassium two pore domain channel subfamily K member 4 - KCNK4. Wide mouth, bitemporal narrowing, bushy and straight eyebrows, long eyelashes, low-set ears, deep/short philtrum, everted upper lip, prominent upper and lower vermilion, micrognathia, and retrognathia are common craniofacial traits. [11].
Conclusion
A genetic syndrome with hypertrichosis, gum hypertrophy, hypoplasia/ aplasia of nails and coarse facial features has a spectrum of disorders. A subtle difference among them helps to differentiate clinically.
References
-
Castori M, Valiante M, Pascolini G, Leuzzi V, Pizzuti A, et al. (2013) Clinical and genetic study of two patients with Zimmermanne Laband syndrome and literature review. Eur J Med Genet 56(10): 570-576.
-
Mallela MK, Moogala S, Polepalle LT, Reddy VC (2013) Zimmermann-Laband syndrome: A rare case report of father and son. J Dr NTR Uni Health Sci 2(2): 147-149.
-
Gripp KW, Smithson SF, Scurr IJ, Baptista J, Majumdar A, et al. (2021) Syndromic disorders caused by gain- of-function variants in KCNH1, KCNK4, and KCNN3-a subgroup of K+ channelopathies. Eur J Hum Genet 29(9): 1384-1395.
-
Megarbane A, Ali R, Choucair N, Lek M, Wang E, et al. (2016) Temple-Baraitser Syndrome and Zimmermann- Laband Syndrome: one clinical entity? BMC Med Genet 17(1): 42.
-
Sheppard SE, Rivera F (1993) Wiedemann-Steiner Syndrome. In: Adam MP, et al. (Eds.), Gene Reviews University of Washington, USA pp: 1993-2024.
-
Deardorff MA, Noon SE, Krantz ID (2005) Cornelia de Lange Syndrome. In: Adam MP, et al. (Eds.), GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle pp: 1993-2024.
-
Demir S, Gurkan H, Oz V, Yalcintepe S, Atli EI, et al. (2021) Wiedemann-Steiner Syndrome as a Differential Diagnosis of Cornelia de Lange Syndrome Using Targeted Next- Generation Sequencing: A Case Report. Mol Syndromol 12(1): 46-51.
-
Lovelace PD, May LA (2023) Nail-Patella Syndrome. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
-
Okamoto N, Ehara E, Tsurusaki Y, Miyake N, Matsumoto N (2018) Coffin-Siris syndrome and cardiac anomaly with a novel SOX11 mutation. Congenit Anom (Kyoto) 58(3): 105-107.
-
Schrier SA, Bodurtha JN, Burton B, Chudley AE, Chiong MA, et al. (2012) The Coffin-Siris syndrome: a proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet A 158A (8): 1865-1876.
-
Elhossini RM, Sayed IM, Hellal US, Mahmoud SAM, Aglan MS, et al. (2024) A recurrent KCNK4 variant in a dominant pedigree with hypertrichosis and gingival fibromatosis syndrome: Variable phenotypic expressivity and insights on patients’ dental management. Am J Med Genet A 194(1): 39-45.
- Epithelioid Granuloma; 3cases with Different Clinical Features
- Advancing Representation in Dermatology Clinical Trials: Ethical, Scientific, and Regulatory Imperatives for Inclusion Across all Fitzpatrick Skin Types
- A Case of Atopic Dermatitis with Concurrent Psoriasis Vulgaris: Successful Treatment with Upadacitinib
- Innovation Lifting Eyeshadow: A Synthesis of Makeup and Optical Illusion
- Distinguishing Superficial Actinic Porokeratosis from Actinic Keratosis with UVF Dermoscopy: A Case Report
- High Mobility Group Box 1 (HMGB1) in Cutaneous Inflammation: An Immune Modulator Bridging Cellular Stress, Ferroptosis and Danger Signaling