Epigenotoxicity of Endocrine-Disrupting Chemicals Makes Inroads to a Paradigm Shift in the Risk Assessment of Pesticides
The most ferocious chronic effect a pesticide could have is disrupting the functions mediated by the endocrine system. When this happens during early development of the life cycle, it leads to profound and lasting adverse human-health and wildlife effects. These effects may not only stop at interrupting some plain physiological functions, but could also play havoc with the epigenetic machinery regulating DNA-transcription in exposed individuals. Even worse is that this havoc and its supervened epigenotoxic effects could be stably transmitted to descendants without further exposing them to the same pesticide or changing their ancestral DNA base sequences. In this manuscript the author reviews the literature with the aim of building a case around the expandable risk posed by Endocrine-Disrupting Pesticides (EDPs); a risk beyond their traditionally-assumed or regulatory-promulgated safety measures. The fact that some EDPs elicit their endocrine-mediated effects at doses far below their acceptable daily intake (ADI) destroys our confidence in the current safety thresholds of human exposure and environmental contamination proclaimed for these pesticides. It seems that the precautionary principle of ‘reasonable certainty of no harm’ for EDPs is only a lip-serviced phrase that has no practical value, especially in long terms, due to the possible transgenerational epigenotoxicity caused by these pesticides. The most important message of this review is that the effects of EDPs and their possible epigenetic inheritance significantly amplify the negative impacts and health hazards of these pesticides and require a paradigm shift from the traditional risk assessment approach to a one that includes evidence-based epigenotoxicology (EBE).
Review
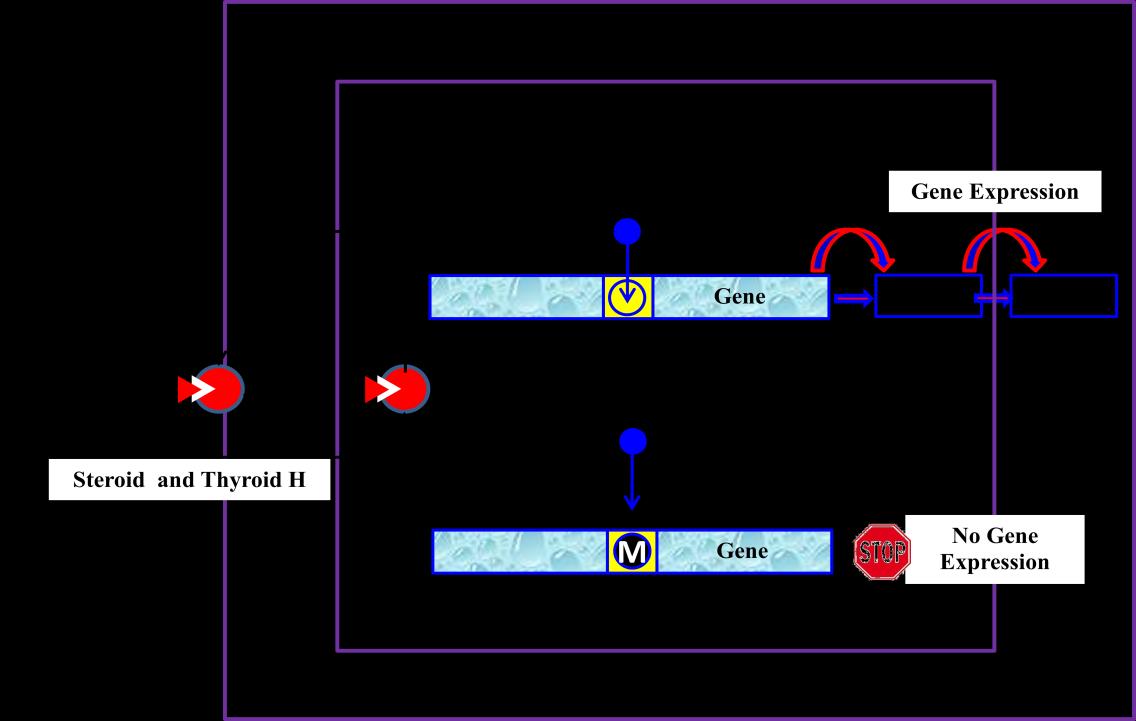
Given the fact that 17,000 publications dedicated or related to the topic of epigenetics appeared only in 2013, with 45 publications a day [12], it is highly difficult or even impossible to make this review a wholly or perfect one. Besides, the field of epigenetics is full of metaphorical language for many reasons. One of the main reasons is that although epigenetic effects are widely- proven, their causal ‘epigenome’ is still indistinct, at least in the physical sense. Therefore only solid and concrete findings will be discussed to possibly serve the objectives of this review in a simple and straightforward way from within a very complicated subject. Metaphoric language will also be used sometimes as a candle to infiltrate darkness in the world of EDPs. For the sake of a systematic and intelligibly-informative flow, it seems appropriate to start this review with a synoptic overview on the endocrine system and how it functions under normal conditions before looking at the possible ways and consequences of its disruption. The Endocrine System: Basic Features and Some Definitions In order to understand the endocrine- disrupting processes and mechanisms; one must first understand the well-known or textbook features of the endocrine system and some related definitions. The endocrine system is similar to the nervous system in that they both are extremely vital communicators in the human body. The means or messengers of communication in the endocrine network are hormones that signal their messages through some specified protein receptors and DNA transcription promoters. In the following, some definitions of the major functional elements of the endocrine system are given. The Endocrine System As defined by the WHO/UNEP [2], the endocrine system is: “a series of ductless glands that secrete hormones directly into the blood to regulate various body functions.” These glands synthesize, store and internally secrete hormones into the bloodstream to be eventually and appropriately delivered to cells, tissues and organs. Endocrine hormones enter cells through the blood vessels to regulate vital functions; such as homeostasis, metabolism, cell differentiation, growth, development, reproduction, response to the environment, especially stressful or injurious stimuli, etc. [13, 14]. In order to function properly, these glands should precisely and timely produce the right amount and deliver it to the right cell/tissue at the right time. The endocrine system is all about temporal- and spatial-signaling of the developmental biology needs to the epigenome and then to the genome to possibly meet these needs. Under normal conditions, the ‘crosstalk’ between the endocrine system, the epigenome and the genome is exquisitely orchestrated in a fine-tuned symphony of DNA expression waves that meets normal development. This symphony of gene expression is delicate and vulnerable to any dissonance caused by, or coming from, EDCs or EDPs with a variegated outcome from resistance/tolerance to acceptance. This is why any chemical that plays minor havoc with our endocrine system can cause major damage to our health, and even to the health of our future generations as will be discussed later. The physiological and developmental needs in our body are communicated to functional systems, tissues and/or organs through message carriers, i.e. the hormones. Hormones The following broader definition is a modified version based on the traditional definition of Melmed & Williams [15] and the one reported by the WHO/UNEP [2]. “Hormones are endogenous chemical molecules produced by specialized cells in a large variety of glands and/or tissues and travel through the blood to produce molecular, biochemical, and physiological effects at sometimes distant tissues or organs and may eventually affect the thinking, feeling and acting of the individual in healthy or unhealthy ways.” In order to transfer the specific messages that convey our ‘regular’ or ‘coping’ needs to our biology-serving and life-supporting systems, hormones must first bind to their specific protein receptors. Hormone Receptors According to Wikipedia [16] “A hormone receptor is a receptor molecule that binds to a specific hormone. Receptors for peptide hormones tend to be cell surface receptors (built into the plasma membrane of cells, thus trans membrane), whereas receptors for steroid hormones are usually found within the cytoplasm (nuclear receptors). The belong to the same definition. Upon hormone binding, the receptor can initiate multiple signaling pathways which ultimately lead to changes in the behavior of the target cells.” The author will use this definition as a basis to structure the functional model of hormones illustrated in Figure 1 (below). Hormone receptors are specialized proteins with special configurations which guarantee high affinity, specificity and effectiveness of their complementary or corresponding hormones. These receptors are always limited in their abundance and are not found in all cells or at all the time. Most hormones also do not act at all times during the life cycle, and their action could be different from one cell type to the other. Even in the same cell type, their action could be different based on the developmental state/stage/age of the organ or organism. In general, there are two types of hormone receptors: membrane receptors and nuclear receptors. Most hormones act through a single receptor and few (some thyroid hormones) act through multiple receptors. The mechanism by which a hormone functions through its receptor(s) is highly complicated and probably not yet entirely disclosed. Therefore, a symbolic and self- explainable representation of this mechanism is shown in Figure 1.
The work of developmental biologist Conrad Hal Waddington in the early forties and that of David Ledbetter Nanney in the early and late fifties has bridged the historic gap between two supposedly inseparable fields, i.e. Developmental Biology and Genetics by means of what was, and still is, called epigenetics or epigenome [12, 17]. Epigenetics explains why a conservative/constant genotype is giving rise to several phenotypes even among identical-DNA-twins [18]. Because Waddington and Nanney independently coined the term epigenetic or epigenetics, they apparently used this term from two different perspectives [19, 20].
Waddington prophetically surmised epigenetics as a means for studying and understanding the ‘causal mechanisms’ by which genes of the genotype interact with the environment and bring about development and phenotypic plasticity. Although Waddington’s perspective was shifted towards developmental biology with a lack of explicit focus on the inheritance of any particular phenotype(s), we consider it to be a revolutionary landmark in a time where DNA- transcription/translation/expression was not in vogue. Nanney, on the other hands, was the one who emphasized that the expression patterns or states of genes could persist through cell division in what is now called ‘cellular heredity or cellular inheritance’, i.e., cell with the same genotype may not only manifest different phenotypes, but phenotypic differences may also persist indefinitely during cellular division in essentially the same environment. These two perspectives had significant impact on the direction of this field till date. However, Nanney should be highly credited for igniting a ‘paradigm shift’ from the ‘Orthodox Heredity’ of Gregor Johann Mendel towards what is now close to an almost credible fact: “not everything that is apparently inherited is necessarily laminated in the genome.” It is even acceptable and safe now to say that our genome should not be treated as an ironclad code of our life; to the contrary, epigenetic malleability allows us to sometimes depart from this code, regardless of whether or not this departure is for the sake of our health and life.
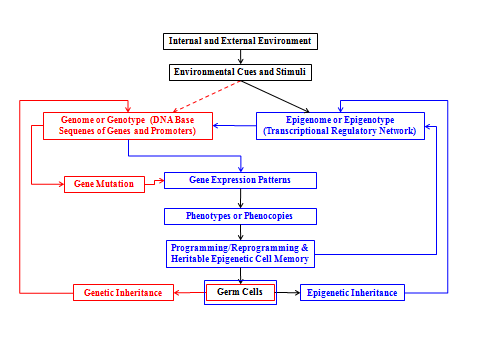
The Waddingtonian and Nanneyan perspectives have been loosely unified by a common interest of understanding how any constant genotype produces different phenotypes [21]. This unification can ‘simply’ be explained by stating that the process of making different phenotypes from the same ancestral genotype cannot happen in regular or irregular development in the absence of a DNA-expression modulator(s); in this case it is the epigenome. Figure 2 (below) is conceptually drawn by integrating our simple perspective with: (1) the hypothesis that the epigenome interfaces environmental cues or environmental information and genomic DNA blueprint to establish transcriptomic profiles and functional identities of individual cell types [22]; and (2) the Holliday’s proposal that epigenetic effects or defects in germ line cells could be inherited in offspring [23]. Figure 2 creates a “Waddington-Nanney-Holliday” recombinant perspective, and also shows that the environmental stimuli or cues play a critical role in phenotypic plasticity through the epigenomic or epigenetic malleability. It is therefore the epigenome’s responsibility to respond to environmental cues and regulate the capability of an organism to adapt and evolve into different phenotypes [10].
Many environmental cues have been found to affect the epigenome including diet, smoking, child care, and environmental pollutants, especially those with endocrine disrupting capabilities [24]. (Figure 2) implies that we are not bound to our genes; in fact, environmental chemicals, especially EDCs and EDPs, could inflict human health and the environment with severe epigenetically- (not just genetically-) mediated adversities. Even worse is that these adversities could be epigenetically transmitted to progenies that are not exposed to these EDCs or EDPs. This phenomenon is called ‘Epigenetic Transgenerational Effects’ or ‘Transgenerational Epigenetics’ or Epigenetic Inheritance’. Hormones are genuinely considered to be the signals that promote specific cell-memorized actions, especially when they exert their effects in the fetal stage and sustain these effects by influencing or dictating the functions of endocrine and physiological systems in later stages or later generation(s).
At this juncture, the meaning of some terms such as epigenome, epigenetic, epigenotype, ‘epigenetics’ and alike should be wrapped up. Although these terms are spreading like a virus in the literature and media during the last two decades, they are diversely ill-defined, and their functions are explained in a rather metaphorical language. It is worth-mentioning that the word ‘epigenetics’ is mistakenly used in the plural form, when it is really singular because it is a term, function, or functional term. It may be appropriate here to sum up this section with two important definitions of the term ‘epigenetics’. Wu and Morris [25] built on many of the previously-debated definitions and stated that “Epigenetics is study of changes in gene functions that mitotically and/or meiotically heritable and that do not entail change in DNA sequence.” It is the author’s opinion that the word ‘function’ be replaced by the word ‘expression’ in the above definition. The justification for such a replacement is rather simple; it is because a change in gene expression can happen without leading to a significant change in its overall function; yet it is still qualified to be named after the term epigenetics. The author also adds his own definition in order to show that the epigenetic machinery is not a sin that ‘only’ causes harm to human-health and the environment; to the contrary, the same machinery can offer means of adversity curing.
The author’s definition states that:“Epigenetics is the machinery by which genes and/or their promoters the disrupted epigenetic processes involved in the initiation and/or progression of malignant cells and other pathophysiological conditions [9].
are marked by physical or chemical tags that without any change in their base sequences could be read by cells to instruct their DNA expression patterns and functions in ways that: (1) meet the temporal/spatial physiological needs of cells during differentiation and development; (2) cope with environmental stress and diseases with adaptation strategies; (3) fail to cope with the environment and experience some health defects; (4) can be recorded, remembered and inherited by cells in a ‘non Mendelian’ heredity fashion.” The bad news is that epigenetic memory of adverse human-health outcomes may lead to transgenerational inheritance of adversity. The good news, however, is that because our epigenome is malleable, some of these memories can be erased after few generations and return to normal states. The study of complex epigenetic mechanisms becomes instrumental not only in understanding the etiopathology of many diseases but in designing their diagnostic markers and therapeutic treatments. Recently, there have been some advances in developing novel drugs (epidrugs) that target The epigenetic machinery and its related tools have been the subject of thousands of research programs, and hundreds of review articles and databases [26]. Because they are not within the scope of this review, only cytosine methylation will be explained as an epigenetic tool to model the effects of EDPs. DNA methylation seems to be the most studied epigenetic regulatory tool of gene expression. It involves marking the genome with methyl (CH3) tags that do not change the DNA base sequences but change their expression patterns or profiles. It was strongly believed that DNA methylation/demethylation reactions take place in the gene promoters within a CpG rich region, now called the ‘CpG islands’. The term CpG refers to the cytosine (C) and guanine (G) bases linked together by a phosphate bond (p) in the DNA nucleotide sequence. Recent studies, however, indicate that methylation can occur equally in cytosines within dinucleotide sequences other than CpGs in undifferentiated cells [27, 28]. DNA methylation is a tool by which a methyl group (CH3) binds covalently to the fifth position of the pyrimidine ring of cytosine predominantly located within the 5´-CpG-3´dinucleotide sequences [29, 30]. Due to its unique chemical and functional identity, the methylated cytosine is sometimes referred to as the 5th nucleotide. Generally, but not always, DNA methylation is associated with gene silencing, i.e., loss of gene expression [31] and vice versa. The CpG methylation is likely evolved as a host-defense mechanism to silence foreign DNA sequences [32]. Most CpG dinucleotides in the human genome are sporadically methylated, while unmethylated CpGs are usually clustered together in the ‘CpG islands’, which are the promoter region that controls the activation/repression processes of gene transcription [31, 33]. Interestingly, the epigenetic marking of the human genome by DNA methylation is heritable (from one cell to the other during cell division), and also stable or persistent through recorded cellular epigenetic memory [34] that may subsequently be transmitted to future generations. Endocrine Disruption: A Potential Mediator to Epigenetic Modification In the first IPCS document [1], an endocrine- disrupting chemical (EDC) was defined as “A potential endocrine disruptor is an exogenous substance or mixture that possesses properties that might be expected to lead to endocrine disruption in an intact organism, or its progeny, or (sub) populations”. A good thing about the IPCS definition is that it implies the essence of epigenetics. However, the definition of Kavlock et al. [35] is also acknowledged in this review for its comprehensiveness. They defined EDC as: "an exogenous agent that interferes with the production, release, transport, metabolism, binding, action or elimination of natural hormones in the body responsible for the maintenance of homeostasis and the regulation of developmental processes." Should the last definition include the effects on ‘(sub) populations and progeny’, it would satisfy the epigenetic effects of EDCs. In an attempt to make a marriage between these two definitions, one can reach the following definition which appears to be more appropriate, especially in the wake of the epigenetic revolution: “EDC is an exogenous substance or mixture which interferes with the synthesis, secretion, transport, binding, action, and/or elimination of natural hormones and eventually leads to disrupting the management and maintenance of homeostasis, reproduction, development and behavior of the intact organism, its progeny, or (sub) populations.” An endocrine-disrupting chemical (EDC) could generally work in many different ways [36, 37, 38] that can be assembled under two major mechanisms. First, an EDC could inhibit or stimulate the synthesis of the hormone or its delivery, with the result that the blood levels of that hormone unjustifiably decline or rise and mismatch the ‘biological necessity waves’ of the hormones-mediated normal functions. This mechanism targets a protein(s) involved in hormone production (e.g. aromatase), an important transporter (e.g. sodium/iodide symporter), or a carrier protein (e.g. cortisol binding protein). The impact of such hormonal disturbance would likely be similar to the situation of diseases or genetic defects that inhibit or stimulate hormone biosynthesis. Second, an EDC could interact ‘directly’ with a hormone receptor in an antagonistic or agonistic manner. The effects could be quite complex in this case, but expected to follow some of the kinetic characteristics of how natural hormones interact with their specific receptors.
ED generally represents a unique kind of toxicity wherein a disruptor is an exogenous chemical that disturbs the endocrine-mediated network and epigenetic manifestations; thereby causing a multitude of adverse human-health and wildlife effects. The mechanism and sequence of events following ED have huge impact on the pattern of effects one would observe in experimental systems (in vitro and in vivo) and in human and wildlife epidemiology. Although the exact mechanistic action(s) of EDCs on reproductive systems is not thoroughly or entirely known, Del-Mazo et al. [39] reviewed several reports and concluded that EDCs are detrimental to these systems in mammals and other species because of their ability to promote abnormalities in sex differentiation and gonad functions, including testicular cancer in the male and ovarian diseases in the female. Examples of the environmental EDCs that have been postulated to elicit adverse effects on the reproductive system in animals and humans are: insecticides (e.g., methoxychlor), fungicides (e.g., vinclozolin), and a range of xenoestrogens (see [39] for review). Reproduction is highly different from growth and development in the types of cells that can be intoxicated by EDCs. As has been clearly stated [40], germ cells are unique as they give rise to a new organism and transmit genetic information to next generations; thus their response to EDCs could be different from that of somatic cells. For example, while in somatic cells epigenetic changes make gene-expression programs progressively more restricted through successive differentiation pathways, germ cells retain intrinsic totipotency. Pesticides and Epigenetic Effects A growing body of scientific evidence shows that humans, domestic animals, wildlife and fish have been experiencing adverse health consequences mediated by ED; as a result of their exposure to environmental chemicals, including pesticides. Some pesticides are known for their endocrine disrupting potential (see [41] for review). These pesticides are referred to as EDPs. In general terms, EDPs are considered to be external cues causing perturbation in the endocrine system and ultimately posing harm to all organisms that rely on this system as one of the most important element in their world of biological information network. It is surprising that decades after the rumors that pesticides could have an ED potential, a consensus about their exact mode of ED action and their acceptable risk assessment protocols, guidelines, methodologies and endpoints remains elusive [42] to a great extent. Many problems, however, have been detected in domestic or wildlife species exposed to some organochlorine compounds (e.g., 1, 1, 1- trichloro-2, 2-bis (p-chlorophenyl); some polychlorinated biphenyls (PCBs), some dioxins; and some naturally occurring plant estrogens [43]. Environmentally-polluting chemicals, such as pesticides, can induce changes in DNA methylation in adults, influence their susceptibility to different pathologies and propagate diseases decades later in their offspring that were only exposed during prenatal and early life [44].
It is the author’s belief that EDPs are much more dangerous than just having an endocrine-disrupting potential. They are likely imposing serious noise into the pathway and network harboring the symphony of gene expression. Unfortunately, the outcome of such a noise could be programmed in the cellular memory and lend itself to transmission to sister and daughter cells within the exposed organism and its descendants. It is generally accepted that environmental factors, particularly those chemicals with an ED-potential are capable of promoting phenotypes with disease states not only in exposed individuals but also in their progeny for some successive generations through epigenetic or transgenerational inheritance [45-47 and references therein]. These environmental factors may not have the potential or the capacity to alter the tenacious genome and promote genetic mutations in the DNA base sequences; but they certainly have the capacity to alter the epigenomic/epigenetic ability of gene expression. Different from the genome, our epigenome is not an ironclad code; to the contrary it is malleable and changes throughout human life [48]. This may explain why ‘epimutation’ happens at higher frequency rates than mutation [49]; and draws attention to the serious, sometimes hardly-proven, risk associated with pesticides, especially those which may find their way to sabotage the endocrine-mediated information and network system in the human body. EDPs and Heritable Epigenotoxicity The sequential effects of EDCs and EDPs could be summed and named after the new term ‘Epigenotoxicity’ which is widely different from the commonly used term ‘Genotoxicity’. The distinction between these two types of chronic toxicity can simply be understood from recalling the functions of, and functional relationship between, the ‘epigenome’ and ‘genome’. The former currently refers to the intertwined machinery tools that regulate the temporal and spatial functions of the latter. Therefore, although genotoxicity requires mutations at the genome level, epigenotoxicity can intoxicate the animal and its descendants without changing its DNA base sequences. The newly-coined Epigenotoxicity term is, therefore, defined within the scope of ED-mediated epigenetic effects as follows: “Epigenotoxicity is a broad type of toxic hazard caused by an agent through its endocrine-disrupting capacity that eventually upsets the orderly course of gene expression and functions and causes an epigenetic health effect(s) that could be heritable either mitotically across different cells in exposed individuals or meiotically to their progeny and un- exposed descendants without entailing any due change in their DNA base sequences.” The issue of transgenerational or epigenetic inheritance of adverse human-health and environmental effects of EDPs was strongly emphasized when the anti- androgenic, well-known fungicide vinclozolin was given at a single time to mice with testis in a critical period of development. As discovered by Anyway et al. [50], vinclozolin produced an adverse effect on the developing testis that was passed on to the following three generations of mice. This effect was explained to be an endocrine- mediated epigenetic change that was transmitted with high fidelity from one generation to the next via reprogramming of the germ-cell memory. Therefore, when environmental pollutants, such as pesticides, are ‘bio-available’ at the time of sex determination, they may change the epigenetic programming and alter the transcriptomic profiles of the
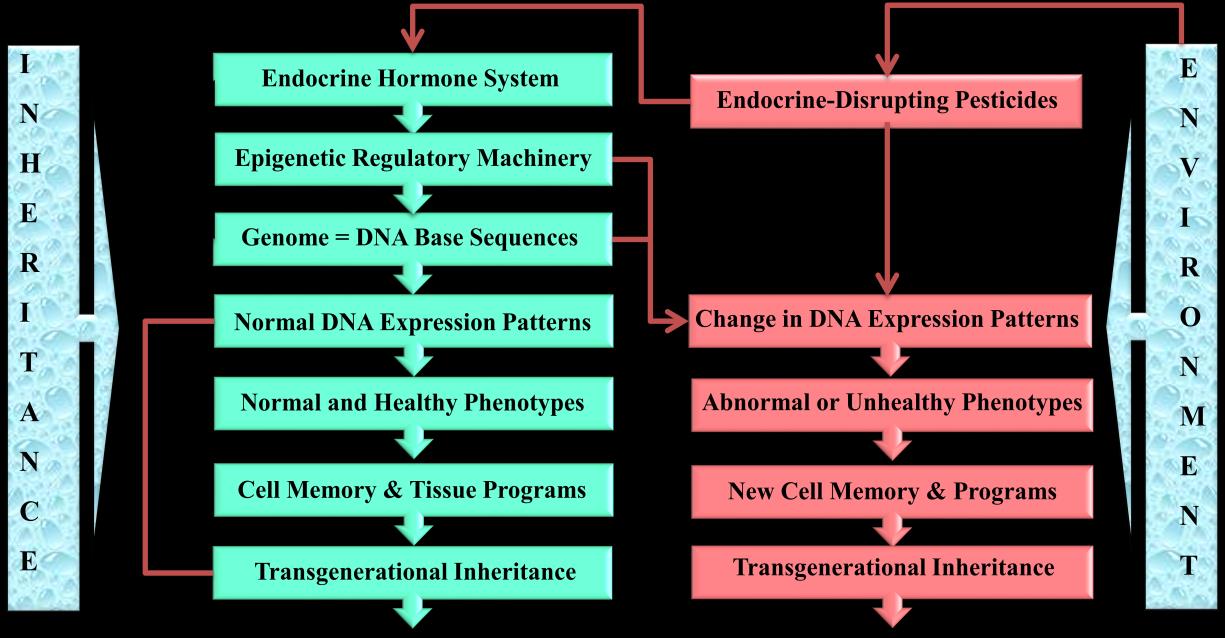
| Figure 3: An illustration shows the proposed sequential effects of EDPs. The endocrine-mediated | |||
| epigenotoxicity may be transmitted to offspring and unexposed descendants. In this review the word | |||
| phenotype(s) is sometimes used when the same individual experiences epigenetic defects during his/her life | |||
| cycle. | |||
| germ line of developing organs and eventually promote | |||
| transgenerational diseases. | |||
| A number of exogenous chemicals have been | |||
| shown to epigenetically influence transgenerational | |||
| mechanisms in animals. Although we have a great deal to | |||
| learn about the epigenetic inheritance of adverse effects | |||
| caused by EDPs, it is plausible that exposures to these | |||
| pesticides during pregnancy will affect the health of | |||
| people from subsequent generations who are not | |||
| themselves exposed to these pesticides. This problem is | |||
| proliferated in rural area with intensive cropping and | |||
| heavy use of agricultural pesticides, wherein the chance of | |||
| exposure is higher than urban cities. Based on | |||
| theprevious review, Figure 3 was designed to illustrate | |||
| how the endocrine system is mediating epigenotoxicity of | |||
| pesticides. This figure shows how the normal profile | |||
| (signature) of gene expression pattern can be changed in | |||
| the presence of EDPs and a new signature, along with | |||
| associated health defects, can be transmitted to next | |||
| generations without the need for genomic mutation. | |||
accumulate and transform normal cells to invasive or metastatic tumor cells [54]. Recent findings show that persistent environmental pollutants, including pesticides, that increase the risk of several types of cancer may in fact act through epigenetic mechanisms ([30] and references therein). In their publication [55], Knower et al. highlighted the epigenetic alterations induced by exposing early-life to EDCs and where possible described how these alterations cause endocrine-related diseases, including breast cancer. In fact all the systems controlled, mediated or modulated by endocrine hormones in our body can be derailed by EDCs and EDPs. The ED- sensitivity window for most endocrine-dependent organisms is the transition from a fertilized egg into an infant. Within this window the cells begin to divide and differentiate and the tissues begin to grow and develop. A delicately fine-tuned status of hormone titers and protein biosyntheses must temporally and spatially match the molecular, biochemical, and physiological needs during these highly critical events. This explains why a dose of a disrupting chemical may do substantial damage to a developing fetus, while exhibiting no or insignificant effects on the mother. Even worse is that cancerous diseases may invade adults who were not exposed to EDCs or EDPs during their independent life, but were exposed in their prenatal stage. In this regard, the author speculates that the effect of an EDP that has a common site(s) of action in target and non-target organisms could have multi-fold effects on humans due to intoxicating this site first and then making the organisms more vulnerable to ED and related endocrine effects including epigenetic cancers.
It is the author’s hypothesis that EDPs follow Poincaré mathematical principle [56] in their effects on the endocrine system that may lead to cancer. The French mathematician Jules Henri Poincaré in the early 20th century indicated that a small difference in the initial conditions of a phenomenon may produce huge effects in the final phenomenon, and a minor error in the former may induce major error in the latter; therefore, prediction becomes highly impossible. In this regards, we hypothesize that EDPs initially distort the information sent by the endocrine system to the epigenome and through cell memory this distortion spreads like a virus and keeps infecting the epigenome over and over in other cells, especially during tumourigenesis. Given the evidence that epigenetics meets endocrinology in many molecular and biochemical arenas [3] and that some pesticides are proven to be strong suspects of ED, it is reasonable to think that these pesticides might interfere with the germ cell reprogramming through epigenetic mechanisms and promote epigenetic cancers [57].
Obviously, perturbation of epigenetic mechanisms can lead to altered gene function and malignant cellular transformation [58]. There are many intertwined epigenetic processes [9] that provide endocrine disruptors a set of interrelated and highly vulnerable targets to cause epigenotoxicity and induce epigenetic malignancy. Since there are many classes of chemicals, including pesticides, that are now known to disrupt the endocrine system, it is reasonable to hypothesize several ways by which EDCs and EDPs could affect the epigenetic machinery and pose cancerous risk to humans. The worst case scenario, which is likely possible, is that the alteration of DNA methylation induced by a single compound could deregulate several genes and produce diverse phenotypic profiles [59]. The epigenetic effect(s) of the synthetic estrogen diethylstilbestrol (DES), a well- known endocrine disruptor, is a good example of this case. In fact DES-exposure of experimental animals was found to be associated with a range of cancers, malformations of the genital track, and obesity [60]. This was explained as probably related to alteration of DNA methylation patterns [61]. Wong and Walker [62] also indicated that the perinatal exposure to EDCs, and especially xenoestrogens, increased cancer risk by (re)programming the epigenomic machinery via alterations in DNA methylation and histone modification. Possible Mechanisms of EDP-Induced Tumors Statistics shows that there is a growing burden of cancer worldwide. According to the Centers for Disease Control and Prevention (CDC), an estimated 168.1 million years of healthy life are lost because of cancer every year [63]. Today people who die from cancer are more than double those who die from malaria, tuberculosis and AIDs combined [64]. Within these horrible statistics, one cannot exactly disclose all the factors and mechanisms of cancer initiation and progression. For the sake of simplicity, however, the author focuses on ED as resounding causes of cancer, and on two mechanisms that are mostly related to DNA methylation (Figure 4). This section is not meant, in any way, to discredit or incriminate pesticides by ‘blindly’ linking them to cancer development, though the possibility is always there, especially for EDPs. ED does not just involve a single/simple biochemical process that can be rescued by antidotes; it is a disaster messing up the whole network and management system that regulates the flow of biological information between the external environment, the endocrine system and the genome. Based on the information received from the external and/or the internal environment and passed to the epigenome
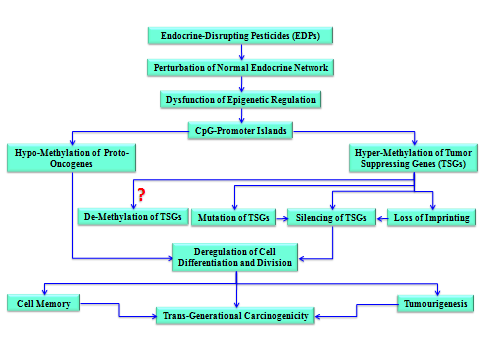
through the endocrine system some instructional/transcriptional decisions are made up by the epigenome to switch the genes on/off and up/down. This delicate and highly instrumental switching machinery is the key to most human diseases, including cancer. Improper or chaotic regulation of epigenetically- mediated DNA expression is common in division and multiplication of human cancer cells (see [9] for review). Carcinogenesis is an outcome of a multistage process involving inappropriate activation of normal genes to become oncogenes [65].
The epigenetic machinery regulating normal and cancerous cell division is using a multitude of biochemical and biophysical reactions [54]. In particular, DNA methylation could be triggered by endogenous or exogenous chemicals that: (a) change the precise dynamics of endocrine signaling patterns, (b) perturb the normal epigenetic guidelines of gene expression, and (c) promote the development of cancerous cells. Because the aberrant DNA methylation appears to be a defining feature in human cancer [9], it is exclusively used to model two mechanisms (the hypo and hyper mechanisms) of epigenetic tumourigenesis (Figure 4). Although they may appear to be different, these two mechanisms, as shown in Figure 4, are basically ED manifestations. The ‘Hypomethylation Mechanism’ is associated with CpG un-methylation or de-methylation (erasure of DNA methylation) that is common in cancer cells [66, 67]. DNA hypomethylation may promote tumourigenesis by transcriptional activation of proto- oncogenes and oncogenic cell growth. The ‘Hypermethylation Mechanism’, on the other hand, is associated with CpG methylation_. The literature is now full of research highlighting the major consequences of epigenetic alterations in cancer cells due to methylation- mediating silencing of the Tumor-Suppressing Genes (TSGs) which are naturally required for regulation of normal cell-growth and differentiation [34]. The hypermethylation mechanism may promote tumourigenesis by proceeding through three different pathways. The first pathway leads to silencing TSGs through the methylation of their CpG-islands and/or those of their promoters. The second pathway that may follow hypermethylation is through the ‘Loss of_ Imprinting’ (LOI). In his commentary article [49], Andrew P. Feinberg indicated the importance of epigenetical LOI in cancer. Regardless of whether this loss is a consequence of methylation or demethylation; these processes are reversible, and we prefer to make it consequent or subsequent to CpG methylation (Figure 4). The third pathway is through mutation of methylated TSGs themselves. The last pathway is supported by the high instability of methylated cytosine (C) and their predisposition to gene mutation through deamination and conversion to thymine (T). Mutating cytosine to thymine could lead to inactivation of TSGs. A simple example of this pathway is when the CGA arginine-encoding trinucleotide mutates to TGA which specifies a stop codon resulting in a premature or truncated protein upon translation [68]. Therefore, one may conclude that the improper epigenetic regulation of CpG methylation/demethylation may function as an endogenous ‘pseudo-mutagen’ that boosts the carcinogenic potential of EDCs and EDPs.
Whereas the three pathways of hypermethylation lead to the same result (e.g., silencing of TSG’s), the first two pathways are reversible; thus enable demethylating agents to remove or erase or scrub the methyl tags and resume TSGs’ expression and stop cancer development. To the contrary, the third pathway is irreversible, at least in the short run, and the literature do not provide supportive evidence for the reversibility of TpG mutated from CH3-CpG. Although acute myeloid leukemia (AML) is characterized by distinct genetic abnormalities, recent discoveries indicated an important role of dysregulated epigenetic mechanisms in its pathogenesis [69]. Being frequently reversible these dysfunctions provided opportunities for targeted treatment of AML using some specific inhibitors.
To show how important epigenotoxicity is, one should know that the epigenetic regulatory machinery that deciphers the information received from the endocrine system upon its reaction with the internal and/or external environment is working through the awakening/silencing of a huge numbers of nucleotides in the mammalian genome. While the coding genes comprise only 1%–2% of the mammalian genomes, some estimates suggest that more than two thirds of these genomes are repetitive or repetitive-derived elements [70]. In the old days, the non-coding DNA was ‘erroneously’ called Junk DNA. The term Junk DNA was popularly used in the sixties but explicitly discussed by David Comings and formalized by Susumu Ohno in 1972 [71]. The activity of non-coding sequences is mostly regulated by epigenetic machinery that is vulnerable to ED. Another complication of epigenotoxicity is that the loss of a temporal and/or spatial homoeostasis of epigenetic regulatory mechanisms may result in perturbation of DNA transcriptional patterns, insertional mutagenesis, and chromosomal aberrations that have been reported in numerous human diseases, including cancer [72]. Epigenotoxic Risk Assessment of EDPs This section deals with the endocrine-mediated epigenetic effects of EDPs from a toxicological viewpoint. The hypothesis that EDPs, either by themselves or jointly with other environmental chemicals, can reprogram the germ line and induce epigenetically transgenerational diseases is a new paradigm in the fields of mammalian toxicology. New fields of ‘Mammalian Epigenotoxicology’ and ‘Epigenotoxic Risk Assessment’ must be inaugurated to fairly study endocrine disruptors. This hypothesis is reasonably justified by a library of findings on many EDCs and few EDPs. Within this hypothesis, one would expect that pesticides that directly or indirectly modulating the endocrine system and its related functions could have ‘actual’ safety exposure thresholds way below the ones promulgated by agrochemical companies and pesticide regulatory authorities. This section brings awareness of the danger of relying on traditional risk assessment protocols, methodologies and endpoints to evaluate the actual risk of EDPs. In their 2015 review with 314 references, Vivian Futran Fuhrman and her colleagues [73] compiled and discussed the uncertainties and unknown that regulators may face when considering the risk assessment of EDCs, including EDPs. Their review indicates clearly that there is no definitive risk assessment tool for these chemicals; a situation that will enforce regulators to accept data from loosely designed testing protocols and poorly defined, even distant or irrelevant, endpoints.
The premise that EDPs can be evaluated for their human and environmental risk in the same manner as non-endocrine-disrupting pesticides is invalid for at least six reasons. First, the traditional approaches and protocols of risk assessment carry the ancillary assumption that tests at high doses will inform us about low-dose exposures. In the light of non-linear or even the non-monotonic dose response (NMDR) relationship of endocrine disruptors [74, 75, 76], any hypothetical extrapolation from higher doses to lower doses is certainly not acceptable with EDPs. The phenomenon of non-monotonicity associated with hormone action/inaction implies that the traditional risk assessment guidelines and methodologies used with all pesticides will restrict the endpoints considered adverse to those mostly unrelated to ED and its manifested epigenotoxicity. In this respect, regardless of the reality and/or the importance of NMDR, risk assessment of EDPs should not be paralyzed by waiting to understand the biological mechanism of this phenomenon or by the challenge raised by traditional toxicologists who are non specialists or experts in the endocrine system, physiological events and the epigenotoxic endpoints of interest. The challenging debate is and should be welcomed within the risk assessment domain of these dangerous chemicals. Second, with the NMDR phenomenon associated with EDCs and EDPs, it is highly difficult, if not impossible, to determine the ‘No Observed Adverse Effect Level’ (NOAEL) or the ‘Lowest Observed Adverse Effect Level’ (LOAEL) when multiple epigenetic endpoints are examined in different in vitro or in vivo models or different species of experimental animals. If this is the case, the estimated exposure thresholds, safety margins, and MRL or tolerance values of EDPs are neither worthy nor trustworthy. Third, the NMDR phenomenon occurs in many levels of toxicological investigation such as in vitro, experimental animals, and human study; on many hormone systems that are targeted by EDCs; and at multiple stages of life cycle [76]. The complexity of these levels and others should not be neglected when studying the risk of EDPs. Fourth, we are talking here about a molecular and biochemical targets (endocrine-mediated epigenetic functions) that are expected to be different from one moment to the other, from one tissue to the other, from one state/stage/age to the other, from one situation to the other, etc. Under such conditions, any extrapolation to calculate the Chronic Reference Dose (cRfD) or the Acceptable Daily Intake (ADI) of EDPs will lead to meaningless or misleading numbers in the least. Fifth, if the fidelity of man-like models from among placental mammals, even the presumed closest human relative (e.g., the old-world macaques primate), is acceptable for traditional toxins, it is a ‘fallacy or myth’ in the least for endocrine disruptors and epigenotoxic agents including EDPs. The conclusion of ‘high fidelity fallacy’ was prophetically reached more than 50 years ago by Russell and Burch [77]. Today, this conclusion is more important than ever before from toxicological and humane perspectives. The huge differences of sensitivity among different animal species and also among different individuals of the same species, makes the extrapolation from experimental animals to humans rather dangerous. If model animals are used, the extrapolation to humans should only be used or taken seriously when the evidence is positive. When experimental animals do not endocrinologically or epigenetically respond to an environmental chemical, the conclusion of no effect should not be extrapolated to imply a reasonable certainty of no harm to humans. With EDPs, the author of this review believes that in vitro human cell testing, methylated reporter and other epigenetic marker assays [78] could be better than, or complementary to, experimental animals with their mushroom or unknown physiological status. This will take us close to Bill Russell and Rex Burch’s principles of humane experimental technique [77, 79]. Russell and Burch’s pivotal publication recognized that the future risk-assessment testing would lie in the use of human cell cultures. Sixth, the epidemiological studies are carried mostly after the fact and the evidence could be related back to ancestral exposure rather than from direct insults. This reason is strengthened by the recent study [11] which showed clearly that the epigenotoxic effects of an insecticidal mixture (permethrin + DEET) lasted for three successive generations. The phenomenon of ‘ancestral exposure and descendant response’ will always discredit the short-term risk assessment protocol for any pesticide with any experimental animal(s).
It unfortunate that using the epigenetic endpoints with model experimental animals in the risk assessment of EDPs will be highly expensive for at least the following four reasons: (1) no one endpoint of hormone disruption and epigenotoxicity can effectively act as a surrogate endpoint for all ED-mediated human- health and environmental outcomes; (2) even multiple epigenetic endpoints with one species of experimental animals may not be predictive of human response which requires the use of more than one species in testing; (3) under all cases, a large number of tested animals is required to meet the fact that individuals with the same age may well experience different physiological state; (4) the effects of EDPs could be epigenetically extended from exposed individuals to their successive generations, an assumption that will never be met by the short-term protocols of traditional risk assessment studies. Conclusion The conclusion of this review article revolves around Figure 5. In this figure, the pathway from ED to heritable diseases including cancer is taking place through some key steps. The conclusion will walk us through these steps within a framework of logical ‘cause- effect’ relationships. Obviously, some connections and details in these relationships are not fully proven, especially in the case of human epigenetic response to EDPs.
People pass on their genes to their children via Mendelian Heredity. In so doing, they pass on various traits and features associated with their genetic blueprint. All the genes in a species are referred to as the ‘genome’ of this species. Only upon expression, this genome can provide us with the life we have and live by. Normal development from this perspective is a process of turning this epigenome that defines and controls tissue development by timely and spatially regulating the awakening/silencing dynamics/waves of gene expression. It is important here to note that the term epigenome is only metaphorical and means nothing but ‘above, over or beyond’ the genome. The epigenome to the genome is like the mind to the brain. There is no physical identity for both the epigenome and the mind; yet they both play a crucial role in our life and health.
The epigenetic machinery receives information from the external and/or internal environment via the endocrine system. The relation between the endocrine system and gene expression is well-studied and long-
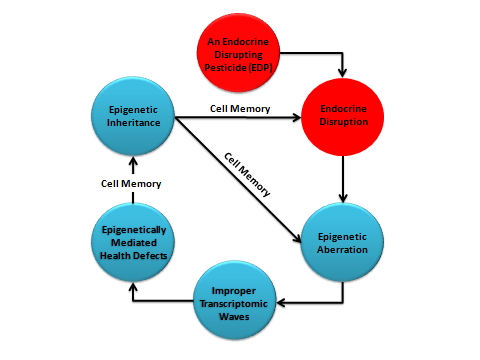
‘on/off’ and/or ‘up/down’ of different combinations of genes, genuinely required for cellular and tissue functions. This simple view of development is not complete without having an apparatus, machinery or operating system ‘above or over’ the genome that is able to ‘differentially’ regulate DNA transcription in due times and places. This operating system is now called ‘epigenome’. Somehow the epigenome manages the exchange and flow of biological information between the environment, our endocrine system and the genome. It is known, with hormones acting as co-regulators of gene expression. Therefore, it is obvious that any agent capable of disrupting the endocrine system will play havoc with the DNA-transcription patterns (transcriptomic profiles). Through unknown mechanism(s), any epigenetic aberration and distorted patterns of gene expression are programmed and installed in the cell memory and transmitted to daughter and sister cells within the exposed organism or its offspring. These epigenetic effects are now known to be heritable without entailing genomic mutation. This is why ‘epigenetics’ is broadly defined as any heritable phenotypic traits independent of any change in the DNA nucleotide sequences. Obviously, one of the most vulnerable sensitivity window(s) to ED is during embryonic sex determination of gonads when the germ line is undergoing epigenetic programming of DNA transcription patterns. In this review, the author argues that EDPs are ‘Epigenetically Active Chemicals’ (EACs) that cause heritable epigenotoxic effects including epigenetic cancer. Depending on the time of human exposure to these pesticides, the epigenetic effects may be transmitted to descendants even without any further exposure.
Although the concept of pesticide-induced disruption of gene-expression is relatively new, one manifestation of ED is to alter a subset of hormone-dependent gene expression; thereby altering development via epigenetic rather than just genetic mechanisms. It is therefore important to indicate that EDPs could be viciously more dangerous than what we think or perceive from their regulatory-promulgated safety thresholds. Their safety measures and risk assessment should go far beyond the study of dose-response relationship that is based on traditional endpoints of health outcomes in experimental animals. A due question before ending this conclusion is: ‘do only pesticides that are known to disrupt the endocrine system play havoc with the dynamics of epigenetically-regulated gene expression and function or do cytoplasmic pesticides also qualify?’ Regardless of any answer to this question, a risk assessment approach based on multiple epigenetic endpoints is long overdue for pesticides. No fixed or single endpoint is reasonably acceptable in EDP risk assessment study, and the data generated from in vitro and experimental animals should be considered with high consciousness and extreme caution.
In the era of epigenetics there are bound to be many changes in regulatory policies for chemicals that enter our environment mostly with good intentions, but may persistently and negatively affect the epigenome of exposed people and their unexposed descendants. The previous sentence contained a message meant to be conveyed to pesticide regulatory authorities worldwide. Nothing is going to danger human life with good intentions as much as the endocrine-disrupting pesticides (EDPs) that are globally used and rampant in our day-to-day life. One example for such phenomenal contamination is that the world is using glyphosate at an annual volume that covers every hectare of global croplands with more than 0.5 kg active ingredient [80]. We share the public and scientific concerns regarding the spread of serious diseases as a result of irresponsible use of pesticides. An example of such a spread – presumably as the result of the use of glyphosate-based herbicides (GBHs) – was recently published [81]. In this study, a correlation between the application of GBHs in the USA and the spread of several human diseases was examined. The authors found a positive and highly significant correlation between annual glyphosate use and the spread of hypertension, stroke, diabetes prevalence, diabetes incidence, obesity, lipoprotein metabolism disorder, Alzheimer’s, senile dementia, Parkinson’s, multiple sclerosis, autism, inflammatory bowel disease, intestinal infections, end stage renal disease, acute kidney failure, cancers of: the thyroid, the liver, the bladder, the pancreas, the kidney, and myeloid leukemia. If the above-mentioned correlation defines a ‘causative’ relationship between glyphosate exposure and its effects, the huge ‘diversity’ among observed diseases indicates that these effects are likely due to epigenetic rather than genetic manifestations of this herbicide. The regulatory impact of the above-mentioned correlation [81] has been seriously discussed in a recent review published by the author of this manuscript [82].
Endnotes and Highlights
• A human being is created with an internal world of biological information; a world that interlinks the external environment, the endocrine system, the nervous system, the epigenome and the genome.
• The epigenome or epigenetics is certainly the theme of the century and is now considered to be the regulating machinery or operating system of our genome.
• Epigenetic machinery relies on some reactions or tools that differentially, temporally and spatially regulate gene expression and functions in waves; thereby determining cell programming and consequent development.
• EDCs and EDPs are those chemicals with a known or suspected perturbing-potential of the endocrine system and related functions.
• Upon ED, a dissonance is introduced into the orchestrated symphony of gene expression and disturbs our life in different molecular, biochemical, physiological, psychological and behavioral ways.
• The area of genetic cancer is now heavily blurred by epigenetic cancer and this puts EDPs on a long overdue ethical and regulatory trial.
• The extended hazard of EDPs can be recorded in cellular memories and epigenetically transmitted to the descendants of exposed individuals without entailing any change in DNA base sequences or any further exposure to these pesticides.
• Due to their complex toxicological nature, the risk of EDPs cannot be based on the traditional assessment protocols, tools and methodologies.
• The most serious toxicological characteristic of EDPs is that their exposure and adverse outcomes are temporarily disconnected, i.e., the 'causative exposure' and 'diseased state' need not to necessarily occur at the same time, the same life stage, or even the same generation; therefore the traditional risk assessment approach significantly undermines or even fails to spot the actual risk of these pesticides.
• With the possible epigenotoxicity, confidence in the traditional safety measures of EDPs promulgated by regulatory authorities is severely damaged or eroded.
• Risk assessment of candidate and already registered pesticides should be revisited and refined using multiple endocrine-epigenetic-mediated endpoints and epigenetic biomarkers.
• The linear and monotonic dose-response (MDR) relationship should not be considered the default in pesticide risk assessment testing; to the contrary the non MDR (NMDR) should be treated just equally in the case of pesticides.
• Because of the likely fraud and conflation of the quality of data, pesticide regulatory authorities should not leave pesticide industry or companies to answer the question of whether: (a) the selected endpoints, protocols and guidelines resolve the risk assessment issues; (b) the species and strains selected are the best choices; (c) the experimental design, including the number of animals, duration of treatment, is justified and well-planned so as to maximize the benefits of these experiments; and (d) the statistical analysis guarantees evidence-based risk assessment measures.
• Collectively, the traditional risk assessment realm of pesticides can be erroneously misleading, especially if these pesticides exhibit an ED-potential.
• It is the author’s opinion that any pesticide 'suspect of ED potential' should be banned regardless of the multiplicity of sources weighing the evidence for such a potential.
• Because the epigenetic risk of EDPs and EDCs is both inevitable and heritable at ultra low exposure levels, integrated pest management (IPM) strategies must be re-invented and seriously implemented.
Competing Interests and Funding
The author declares that he has no competing interests, and does not receive any fund to write this review.
Acknowledgement
It is to firstly acknowledge the great efforts of anonymous reviewers and editors who helped to sharpen the terrain of thoughts in this review. The author wishes to express his deep gratitude to Dr. Salah Abdel Moemen, Former Minister of Agriculture and Land Reclamation, Egypt; Dr. Hossam Ezz Eldin, Dean of the College of Agriculture, Assiut University, Egypt; Dr. Monir D. Abdallah and Dr. Mohamed A. Kandil, Professors of Toxicology, Cairo University, Egypt; Dr. Nagi Abouzid, Professor of Plant Pathology, Dr. Mohsen El-Gendy and Dr. Monir Almaz, Professors of Toxicology, Ministry of Agriculture and Land Reclamation, Egypt for their ever-lasting, friendly and cordial support. Dr. Michael K. Skinner, Center for Reproductive Biology, School of Biological Sciences, Washington State University, Pullman, WA, USA and Dr. Carrie Deans, Department of Entomology, Texas A&M University, College Station, Texas, USA, have been very kind communicating with the author and providing him with some valuable epigenetic literature. Last but far from least, the author acknowledges Mrs. Abla El-Rouby, Mr. Kamal I. Zaki and Dr. Mona K. Ibrahim for their sincere and sustainable support.
References
-
Damstra T, Barlow S, Bergman A, Kavlock R, Van Der Kraak G (2002) Global Assessment of the State-of-the-Science of Endocrine Disruptors. An assessment prepared by an expert group on behalf of the World Health Organization, the International Labour Organization, and the United Nations Environment Programme.
-
WHO/UNEP (2013) State of the science of endocrine disrupting chemicals – 2012: An assessment of the state of the science of endocrine disruptors prepared by a group of experts for the United Nations Environment Programme (UNEP) and WHO; Eds: Bergman Å, Heindel JJ, Jobling S, Kidd KA, Zoeller RT.
-
Xiang Zhang, Shuk-Mei Ho (2011) Epigenetic meets endocrinology. Journal of Molecular Endocrinology 46: 11-32
-
Ankolkar M, Balasinor NH (2016) Endocrine control of epigenetic mechanisms in male reproduction. Horm Mol Biol Clin Investig 25(1): 65–70.
-
Mercola.com (2010) Why Tour DNA Isn’t Your Destiny.
-
Yang O, Kim HL, Weon JI, Seo YR (2015) Endocrine- disrupting Chemicals: Review of Toxicological Mechanisms Using Molecular Pathway Analysis. J Cancer Prev 20(1):12-24.
-
Egger G, Liang G, Aparicio A, Jones PA (2004) Epigenetics in human disease and prospects for epigenetic therapy. Nature 429 (6990): 457-463.
-
Trerotola M, Relli V, Simeone P, Alberti S (2015) Epigenetic inheritance and the missing heritability. Hum Genomics 9:17.
-
Yadav R, Srivastava A, Chandra S, Rai AK (2016) Role of epigenetic mechanisms in various cancer therapies. PHARMACEUTICAL AND BIOLOGICAL EVALUATIONS 3(2): 178-184.
-
Rivera CM, Ren B (2013) Mapping human epigenomes. Cell155(1):39-55.
-
Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM, Skinner MK (2012) Transgenerational Actions of Environmental Compounds on Reproductive Disease and Identification of Epigenetic biomarkers of Ancestral exposures. PLoS One 7(2): e31901.
-
Deans C, Maggert KA (2015) What do you mean, “epigenetic”? Genetics 199(4): 887-896.
-
Hiller-Sturmhöfel S, Bartke A (1998) The Endocrine System: an overview. Alcohol Health Res World 22(3):153-164.
-
Sargis, RM (2016) About the Endocrine System: Endocrine Glands and Hormones.
-
Melmed S, Williams RH (2011) Williams textbook of Endocrinology. Philadelphia, PA, Elsevier/Saunders.
-
Wikipedia (2016) Hormone receptor.
-
Holliday R (2006) Epigenetics: A Historical Overview. Epigenetics 1(2): 76-80.
-
Tan Q, Christiansen L, Hjelmborg JvB, Christensen K (2015) Twin methodology in epigenetic studies. J Exp Biol 218(Pt 1): 134-139.
-
Waddington CH (1942) The epigenotype. Endeavour 1: 18. Republished in International Journal of Epidemiology 1–4.
-
Nanney DL (1958) Epigenetic control systems. Proc Natl Acad Sci USA 44(7): 712-717.
-
Haig D (2012) Commentary: The epidemiology of epigenetics. Int J Epidemiol 4(1): 13-16.
-
Casati L, Sendra R, Sibilia V, Celotti F (2015) Endocrine disrupters: the new players able to affect the epigenome. Front Cell Dev Biol 3: 37.
-
Holliday R (1987) The inheritance of epigenetic defects. Science 238(4824): 163-170.
-
Casati L, Colciago A, Celotti F (2010) Epigenetic mechanisms in health and diseases. Brasília Med_._ 48: 209-218.
-
Wu Ct, Morris JR (2001) Genes, genetics, and epigenetics: a correspondence. Science 293(5532): 1103–1105.
-
Epigenetic Databases, Tools and Resources (2016).
-
Fuso A, Ferraguti G, Grandoni F, Ruggeri R, Scarpa S, et al. (2010) Early demethylation of non-CpG, CpC- rich, elements in the myogenin 5'-flanking region: a priming effect on the spreading of active demethylation?. Cell Cycle 9(19): 3965-3976.
-
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, et al. (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 19; 462(7271): 315–322.
-
Gruenbaum Y, Stein R, Cedar H, Razin A (1981) Methylation of CpG sequences in eukaryotic DNA. FEBS Lett 124 (1), 67-71.
-
Barbara Stefanska, Vinken M, Szyf M (2012) Epigenetics in Toxicology: The Implications of Epigenetic Alterations Driven by External Exposures for Human Health. Altex Proceedings, 1/12, Proceedings of WC8 173-85.
-
Bird AP (1986) CpG-rich islands and the function of DNA methylation. Nature 321(6067):209-213.
-
Hedges DJ, Batzer MA (2005) From the margins of the genome: mobile elements shape primate evolution. Bioessays 27(8):785–794.
-
Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, et al. (1982) Amount and distribution of 5- methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10(8): 2709–2721.
-
Lim DHK, Maher ER (2010) SAC review: DNA methylation: a form of epigenetic control of gene expression. The Obstetrician & Gynaecologist 12: 37– 42.
-
Kavlock RJ, Daston GP, DeRosa C, Fenner-Crisp P, Gray LE, et al. (1996) Research needs for the risk assessment of health and environmental effects of endocrine disruptors: a Report of the U.S. EPA- sponsored workshop. Environ Health Perspect 104 Suppl 4: 715-740.
-
NIH (2010) Endocrine Disruptors.
-
TiPED (2014) What Is Endocrine Disruption.
-
Wikipedia (2016) Endocrine disruptor.
-
Del-Mazo J, Brieño-Enríquez MA, García-López J, López-Fernández LA, De-Felici M (2013) Endocrine disruptors, gene deregulation and male germ cell tumors. Int J Dev Biol 57(2-4): 225-239
-
De Felici M, La Sala G (2016) Epigenetic Reprogramming in the Mammalian Germ Line: Possible Effects by Endocrine Disruptors on Primordial Germ Cells. The Open Biotechnology Journal 10, (Suppl-1, M4) 36-41.
-
Mnif W, Hassine AIH, Bouaziz A, Bartegi A, Thomas O, et al. (2011) Effect of Endocrine Disruptor Pesticides: a review. Int J Environ Res Public Health 8(6): 2265- 2303.
-
McKinlay R (2007) Endocrine disrupting pesticides – more precaution needed. Risk assessment Pesticides News.
-
EPA (2016) What is Endocrine Disruption?.
-
Kanherkar RR, Bhatia Dey N, Csoka AB (2014) Epigenetics across the human lifespan. Front Cell Dev Biol 2:49.
-
Anway MD, Skinner MK (2006) Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 147(6 Suppl): S43-S49.
-
Skinner MK, Manikkam M, Guerrero Bosagna C (2010) Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab 21(4): 214-222.
-
Miousse IR, Chalbot M CG, Pathak R, Lu X, Nzabarushimana E, et al. (2015) _In Vitro_ Toxicity and Epigenotoxicity of Different Types of Ambient Particulate Matter. Toxicol Sci 148(2):473-487
-
NIH (2016) Epigenomics.
-
Feinberg AP (2001) Cancer epigenetics takes center stage. Proc Natl Acad Sci USA 98(2): 392-394.
-
Anway MD, Cupp AS, Uzumcu M, Skinner MK (2005) Epigenetic Transgenerational Actions of Endocrine disruptors and male Fertility. Science 308(5727): 1466-1469.
-
Jones PA, Laird PW (1999) Cancer epigenetics comes of age. Nat Genet 21(2): 163–167.
-
Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3(6): 415– 428.
-
Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, et al. (2015) Executive Summary to EDC-2: The Endocrine Society's Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev 36(6): 593-602.
-
Krupanidhi S, Sedimbi SK, Sanjeevi CB (2008) Epigenetics and epigenetic mechanisms in disease with emphasis on autoimmune diseases. J Assoc Physicians India 56: 875-880.
-
Knower KC, To SQ, Leung, Y-K, Ho SM, Clyne CD (2014) Endocrine disruption of the epigenome: a breast cancer link. Endocr Relat Cancer 21(2): T33- 55.
-
Poincaré H (1903) Computer Viruses and the Human Mind. Science and Hypothesis. Dover reprint, 1952. Mineola, New York: Dover Publications. Cited in: Robertson R (1997).
-
Laird PW (2005) Cancer epigenetics. Human Molecular Genetics 14(1): R65–R76.
-
Sharma S, Kelly TK, Jones PA (2010) Epigenetics in cancer. Carcinogenesis 31(1): 27-36.
-
Anderson AM, Carter KW, Anderson D, Wise MJ (2012) Coexpression of nuclear receptors and histone methylation modifying genes in the testis: implications for endocrine disruptor modes of action. PLoS One. 7(4): e34158.
-
Newbold RR (2011) Developmental exposure to endocrine-disrupting chemicals programs for reproductive tract alterations and obesity later in life. Am J Clin Nutr 94(6 Suppl): 1939S-1942S.
-
Sato K, Fukata H, Kogo Y, Ohgane J, Shiota K, et al. (2009) Neonatal exposure to diethylstilbestrol alters expression of DNA methyltransferases and methylation of genomic DNA in the mouse uterus. Endocr J 56(1): 131-139.
-
Wong RL, Walker CL (2013) Molecular Pathways: environmental estrogens activate nongenomic signaling to developmentally reprogram the epigenome. Clin Cancer Res 19(14): 3732-3737.
-
CDC (2016) Centers for Disease Control and Prevention. Global Cancer Statistics. http://www.cdc.gov/cancer/international/burden.ht m,
-
CDC (2016) Centers for Disease Control and Prevention. The Global Burden of Cancer.
-
Harris CC (1996) p53 Tumor Suppressor Gene: At the Crossroads of Molecular Carcinogenesis, Molecular Epidemiology, and Cancer Risk Assessment. Environmental Health Perspectives 104(S3): 435- 439.
-
Feinberg AP, Vogelstein B (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301(5895): 89-92.
-
Chilukamarri L, Hancock AL, Malik S, Zabkiewicz J, Baker JA, et al. (2007) Hypomethylation and Aberrant Expression of the Glioma Pathogenesis–Related 1 Gene in Wilms Tumors. Neoplasia 9(11): 970 - 978.
-
Rideout WM, Coetzee GA, Olumi AF, Jones PA (1990) 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 249(4974): 1288–1290.
-
Wouters BJ, Delwel R (2016) Epigenetic and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 127(1): 42-52.
-
de Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD (2011) Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PLoS Genet 7(12), e1002384.
-
Wikipedia (2016) Noncoding DNA. Retrieved on June 2, 2016.
-
Miousse IR, Chalbot, M-CG, Lumen A, Ferguson A, et al. (2015) Response of transposable elements to environmental stressors. Mutat. Res. Rev. Mutat. Res. 335, 11–19.
-
Fuhrman VF, Tal A, Arnon S (2015) Why endocrine disrupting chemicals (EDCs) challenge traditional risk assessment and how to respond. Journal of Hazardous Materials 286 (2015): 589–611.
-
Heindel JJ, Zoeller RT, Jobling S, Iguchi T, Vandenberg L, et al. (2013) What is endocrine disruption all about?, Chapter 1, pages 1-22, In: Endocrine Disrupting Chemicals – 2012: An assessment of the state of the science of endocrine disruptors prepared by a group of experts for the United Nations Environment Programme (UNEP) and WHO; Eds: Bergman Å, Heindel JJ, Jobling S, Kidd KA, Zoeller RT.
-
Lagarde F, Beausoleil C, Belcher SM, Belzunces LP, Emond C, et al. (2015) Non-monotonic dose-response relationships and endocrine disruptors: a qualitative method of assessment. Environmental Health 14:13.
-
Zoeller RT, Vandenberg LN (2015) Assessing dose– response relationships for endocrine disrupting chemicals (EDCs): a focus on non-monotonicity. Environmental Health 14: 42.
-
Russell WMS, Burch RL (1959) The Principles of Humane Experimental Technique. London: Methuen & Co. Special edition published in 2016, Johns Hopkins University.
-
Wei J-H, et al. (2015) A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. NATURE COMMUNICATIONS.
-
Goldberg AM (2010) The Principles of Humane Experimental Technique: Is It Relevant Today? Altex 27: 25-7.
-
Benbrook CM (2016) Trends in glyphosate herbicide use in the United States and globally. Environ Sci Eur 28:3
-
Swanson NL, Leu A, Abrahamson J, Wallet B (2014) Genetically engineered crops, glyphosate and the deterioration of health in the United States of America. Journal of Organic Systems, 9(2): 6-37.
-
Ibrahim YA (2015) A regulatory perspective on the potential carcinogenicity of glyphosate. Journal of Toxicology and Health ISSN 2056-3779 | Volume 2 | Article 1.
- Pattern of Gonadal Hormones in Oral Testosterone-Supplimented Male Wistar Rats with Diabetes-Induced Hypogonadism
- Re-Evaluation of the Genotoxicity of Currently Used Food Dyes in Mouse Multiple Organs Via Continuous Administration by Drinking Using the Comet Assay
- Pharmacogenetics of Type 2 Diabetes Mellitus: Linking Genetic Variability to Drug Efficacy and its Cardiovascular Outcomes
- Exploratory Proteomic Profiling of SARS-CoV-2 Infected THP-1 Macrophages Reveals Alterations in Inflammatory Response and Cellular Metabolism
- Study of Genotoxicity of Hepatocarcinogens in Multiple Organs in Mice by Feeding and Drinking Using the Comet Assay
- Spirulina Polypeptides Inhibit the Growth of Human Lung Tumor (H460) Cells