Pharmacokinetics and Bioequivalence of Methylphenidate Hydrochloride 10 mg Immediate-Released Tablets in Healthy Thai Volunteers under Fasting Conditions
Methylphenidate is a central nervous system stimulant indicated for attention deficit hyperactivity disorder (ADHD) and narcolepsy. Methylphenidate GPO® had been developed as a generic alternative to Ritalin® 10 for Thai people. The aims of this study were to characterize pharmacokinetics in Thai population, and to evaluate bioequivalence of Methylphenidate GPO® to Ritalin® 10 to support product registration. A comparative dissolution test was performed in four dissolution media, followed by an open-label, randomized, two-way crossover bioequivalence study under fasting conditions. A single dose of the test or reference product was administered in period I and switched over to another product in period II after 7-day washout period. Blood samples were collected at predefined time points over 24 hours after dosing. Plasma concentrations of methylphenidate were quantified using a validated liquid chromatography-mass spectrometry method. The pharmacokinetics was characterized from plasma concentration-time profile following administration of the test and reference formulations. The pharmacokinetic parameters were in agreement with the previously published data. The AUC0-t, AUC0−∞ and Cmax of two methylphenidate 10 mg tablet formulations were statistically compared in 23 healthy Thai volunteers. The analysis of variance (ANOVA) did not show any significant difference between the formulations. The ratios for geometric least-square means and 90% confidence intervals of log-transformed parameters were within the acceptance range of 80.00–125.00%. Both products were generally well tolerated by healthy Thai subjects. Methylphenidate GPO® and Ritalin® 10 were bioequivalent in terms of rate and extent of absorption, and could be used interchangeably.
Introduction
Methylphenidate is a central nervous system stimulant indicated for attention deficit hyperactivity disorder (ADHD) and narcolepsy [1]. Although the mechanism of action is not well understood, it is possible that methylphenidate increases dopamine levels in the striatum by binding to the dopamine transporter in the presynaptic cell membrane, and consequently blocking reuptake of dopamine [2]. Many studies have demonstrated the efficacy of methylphenidate on improvement of cognitive and behavioral responses in children. Therefore, it has become a first-line therapy for ADHD in children and adolescents [3].
Methylphenidate dosage typically starts with 5 mg twice daily and can be increased up to maximum dose of 60 mg/ day divided 2-3 times daily [4]. After oral administration, methylphenidate is rapidly absorbed reaching maximum concentration between 1 and 2 hours [5]. Although there is no significant food effect, methylphenidate is instructed to be taken 30-45 minutes before meal to minimize risk of toxicity [6]. Methylphenidate shows low plasma protein binding and high lipid solubility, facilitating penetration into effect site in the brain [6]. It is extensively hydrolyzed to inactive metabolite, ritalinic acid. This metabolite is excreted in urine accounted for about 80% of the dose [5, 6]. Terminal half- life is approximately 2-3 hours whereas duration of action is between 1 and 4 hours [4]. Nonlinear pharmacokinetics is observed in the dose range of 10-60 mg [7].
Methylphenidate GPO® had been developed as a generic alternative to Ritalin® 10 for Thai people. The aims of this study were to characterize pharmacokinetics in Thai population, and to evaluate bioequivalence of Methylphenidate GPO® to Ritalin® 10 to support product registration.
Materials and Methods
Study Products
The test and reference products of methylphenidate hydrochloride 10 mg tablets used in this study were Methylphenidate GPO® manufactured by the Government Pharmaceutical Organization (GPO), Thailand (Lot No. S600113), and Ritalin® 10 manufactured by Novartis Farmaceutica, Spain (Lot No. BAK10), respectively.
In Vitro Dissolution Study
The dissolution study was carried out on 12 units each of the test and reference products using the Apparatus II (paddle) method as per the United States Pharmacopeia (USP) monograph. The test was conducted using 1000 mL of dissolution media: water, simulated gastric fluid without enzyme pH 1.2, acetate buffer pH 4.5, and simulated intestinal fluid without enzyme pH 6.8 maintained at 37 ± 0.5°C. The paddle speed was set at 50 rotations per minute (rpm). The dissolution samples were collected at 10, 15, 20, 30, 45 and 60 minutes. Each sample was analyzed by high-performance liquid chromatography (UltiMate 3000, Thermo Fisher Scientific Inc., USA). The mean dissolution values at each sampling time point were used to calculate similarity factor (f2) as following equation:
where Rt is the percentage of dissolved drug for the reference product at time point t, Tt is the percentage of dissolved drug for the test product at time point t, and n is the number of time points used for calculation.
Study Subjects
Total 24 subjects were enrolled in the study. Their ages ranged from 18 to 55 years and their body mass index (BMI) values were between 18 and 30. None of them had underlying disease or abnormal finding during physical and laboratory examinations. In case of female subjects, they demonstrated negative urine pregnancy test and were not breastfeeding women. Females with child-bearing potential agreed to use acceptable methods of birth control including non-hormonal methods and sexual abstinence throughout the course of study.
Subjects were not enrolled or withdrawn from the study if they met predefined exclusion criteria including history of allergic reaction to the study drug or any medications; presence of illness within 4 weeks prior to the start of the study; positive result of suicidal risk assessment; participation in any other clinical trial involving drug administration in 90 days prior to the start of the study; recent history of blood donation or difficulty in venipuncture; recent use of alcohol with positive breath test; consumption of product containing grapefruit, pomelo or orange within 48 hours prior to dosing; consumption of xanthine containing products within 24 hours prior to dosing; cigarettes or tobacco smoking; positive urine screening test for recreational drug use; consumption of any medications, vitamins, or dietary supplements within 14 days prior to dosing. Study Design This study was designed as a comparative open- label, randomized, single dose, two-way crossover study to determine the bioequivalence of methylphenidate hydrochloride 10 mg tablet formulations after oral administration to healthy Thai volunteers under fasting conditions. All the subjects gave written informed consent prior to the start of the study. They were randomly assigned into one of two groups, TR and RT meaning that the subjects either received the test (T) or reference (R) product in period I, and switched over to another product in period II after 7-day washout period. The subjects fasted for at least 10 hours prior to drug administration. One tablet of the test or reference product was orally administered to the subjects with 240-mL water, immediately followed by mouth and hand check to assess the compliance in dosing. Subjects remained in sitting position for at least 2 hours after administration. The adverse events were monitored throughout the study by voluntary report, direct questioning, physical examination and laboratory examination. Severity and causality of the adverse events to the investigational products were assessed and reported to the ethic committee in a timely manner. The clinical study protocol was reviewed and approved by Institute for the Development of Human Research Protections (IHRP), Health Systems Research Institute (HSRI), Thailand on 22 November 2018 (letter no. 798/2561). The study was conducted as per the protocol, ICH Guidance on Good Clinical Practice and Declaration of Helsinki, and Standard Operation Procedures (SOPs) of Clinical Research Center, Medical Life Science Institute, Department of Medical Sciences, Ministry of Public Health, Thailand.
Blood Sampling
Total 20 blood samples were collected from each subject at pre-dose (0 hour), 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 16 and 24 hours post-dose in each period. Approximate 4 mL of each blood sample was drawn from an indwelling cannula in the forearm vein or fresh vein puncture using syringe. The drawn samples were transferred into dipotassium ethylenediaminetetraacetate (K2EDTA)- containing vacutainers and subsequently inverted several time to ensure mixing of tube contents. Approximate 2 mL of each plasma sample was achieved from centrifugation at 3000±100 relative centrifugal force (rcf) for 5 minutes at 4°C. The separated plasma samples were transferred into polypropylene tubes containing around 20 µL of 10% (v/v) formic acid solution (1% of total plasma volume). The plasma samples were vortexed and divided into first lot and second lot for subsequent sample analysis. Samples were stored upright in the freezer maintained at -55°C or colder until analysis.
Sample Analysis and Incurred Sample Reanalysis (ISR)
Bioanalysis was carried out at GPO, Thailand in accordance with the Principles of Good Laboratory Practice, European Medicines Agency (EMA) guideline on the investigation of bioequivalence [9] and in-house SOPs. A 200- µL plasma sample was spiked with 50 µL of internal standard solution, 5 ng/mL of [2H9]-Methylphenidate. Thereafter, 300 µL of methanol was added, followed by vortexing for 3 minutes. Then the sample was centrifuged at 4000±100 rcf for 5 minutes at 10°C. The separated supernatant was transferred into appropriate vial for analysis. Concentration of methylphenidate in the study samples were determined by an ultra-high performance liquid chromatography (NexeraTM, Shimadzu Corporation, Japan) coupled with electrospray ionization triple quadrupole mass spectrometry (TSQ Quantum Ultra, Thermo Fisher Scientific Inc., USA). The vials were placed in an autosampler maintained at 4°C. The samples at a volume of 20 µL were injected into ZORBAX Eclipse XDB-C8 4.6×150 mm column maintained at 40°C.
The mobile phase, 5 mM ammonium acetate buffer pH 3.5: Methanol (40:60, v/v) was pumped at a rate of 0.5 mL/min. Methylphenidate and internal standard were detected in positive ionization mode using a transition of 234.14/84.32 and 243.19/93.34, respectively.
The calibrators and quality control samples were prepared in the screened blank plasma. The weighted (1/concentration2) regression of peak area ratio of methylphenidate to the internal standard was constructed using 8 calibrators covering a range of concentration between 0.1 and 25.185 ng/mL. Four sets of quality control samples at 4 concentration levels (n = 16) covering study sample concentrations were interspersed in each analytical run. The study samples with concentrations below limit of quantification were reported as ‘BLQ’ whereas the samples with concentration above highest calibrator concentration were reanalyzed after dilution with the screened plasma. Data were analyzed using XcaliburTM 4.0.27.42 and LCquanTM 3.0.26.0 (Thermo Fisher Scientific Inc., USA). The analytical method was validated as per EMA guideline on bioanalytical method validation [10] and U.S. FDA guidance for industry: bioanalytical method validation [11]. The analysis was completed within 82 days based on validated long-term stability data.
Study samples having concentrations close to maximum concentration and in the elimination phase of each subject in each period were chosen for incurred sample reanalysis (ISR) in separate analytical runs. According to EMA guideline on bioanalytical method validation [10], at least 10% of first 1000 samples and 5% of the number of samples exceeding 1000 samples were reanalyzed, but not used for pharmacokinetic calculation.
Pharmacokinetic and Statistical Analysis
The pharmacokinetic profiles were established for the test and reference products using respective concentration and time data. Pharmacokinetic parameters were determined by non-compartmental analysis (Phoenix WinNonlin Software Version 6.4, Pharsight Corporation, USA). The maximum plasma concentration of methylphenidate (Cmax) and the time to reach Cmax (tmax) were taken directly from the observed pharmacokinetic profiles. The elimination rate constant (λZ) was determined from the slope of the log terminal elimination phase of pharmacokinetic profiles. The apparent elimination half-life (t1/2) was calculated as 0.693/ λZ. The area under the curve from time zero to last measuring time point (AUC0-t) of pharmacokinetic profiles was calculated by trapezoidal rule. The extrapolated of AUC from last measuring time point (Ct) to infinity was determined as Ct/λZ, thereby using for AUC0-∞ calculation. The AUC0−t, AUC0−∞ and Cmax were reported as primary pharmacokinetic parameters, whereas tmax, λZ and half-life (t1/2) were reported as secondary pharmacokinetic parameters.
The statistical analysis was carried out using PROC GLM (SAS® Version 9.4, SAS Institute Inc., USA). Analysis of variance (ANOVA) was performed for log-transformed pharmacokinetic parameters: AUC0-t, AUC0-∞ and Cmax. ANOVA mixed-effect model included sequence, treatment, period and sequence as fixed effects, and subject nested within sequence as a random effect. Sequence effect was tested using subject nested within sequence as an error term. The significance of these effects was determined using F-test. The two one-sided tests were carried out by computing the 90% confidence interval (CI) for the ratio of the geometric least square mean (test/reference) of log-transformed primary pharmacokinetic parameters, which should be within the acceptance range of 80.00-125.00%. Wilcoxon-signed rank test was performed to compare tmax of the test and reference products. All statistical calculations were performed at a significance level of 5% (α=0.05). Study subjects who did not provide evaluable data for both of the test and reference products were excluded from statistical analysis.
Results
In Vitro Dissolution Study
The dissolution profiles of the Methylphenidate GPO® and Ritalin® 10 in water, simulated gastric fluid without enzyme pH 1.2, acetate buffer pH 4.5, and simulated intestinal fluid without enzyme pH 6.8 are illustrated in Figure 1-4, respectively. The results showed that methylphenidate was dissolved more than 85% of labeled amount within 15 minutes in all tested media. Therefore, the dissolution profiles were accepted as similar without f2 calculation [8].
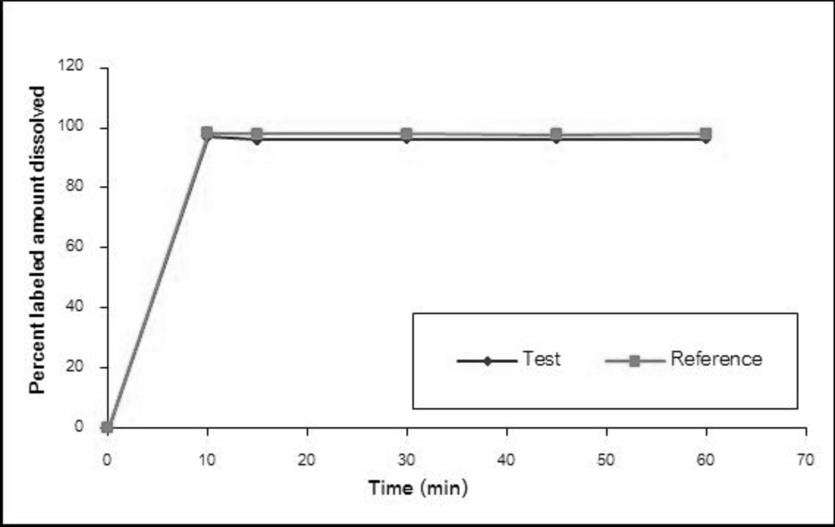
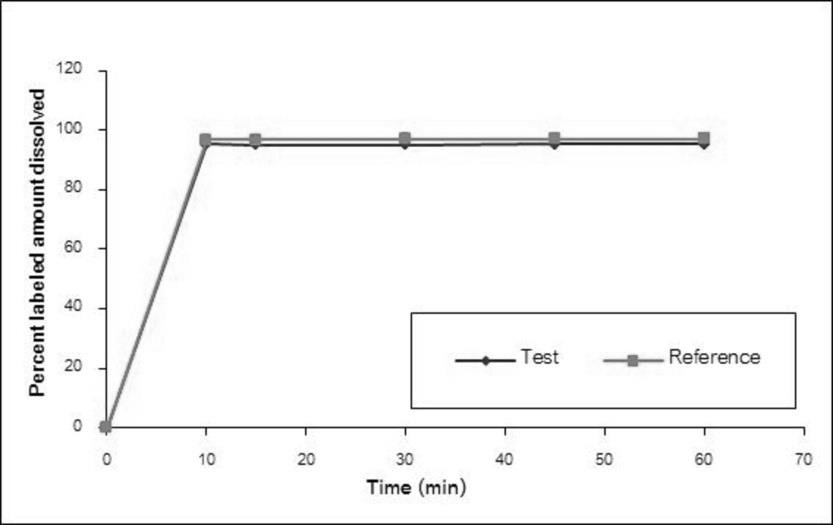
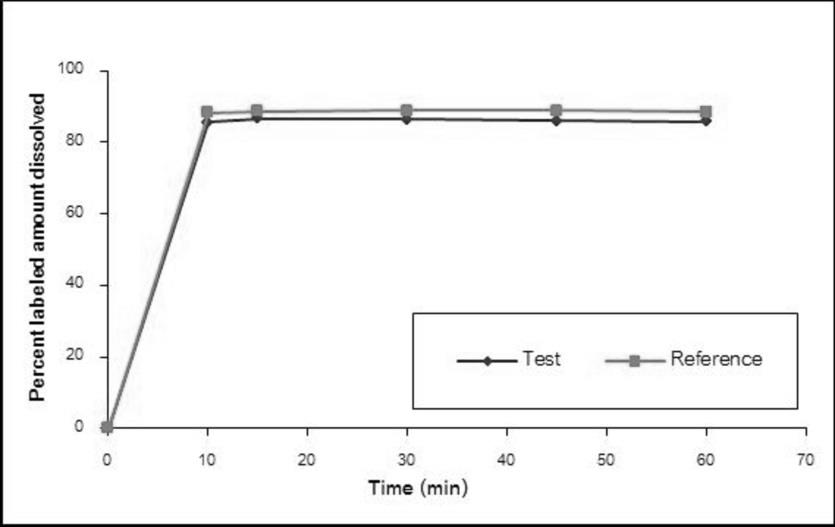
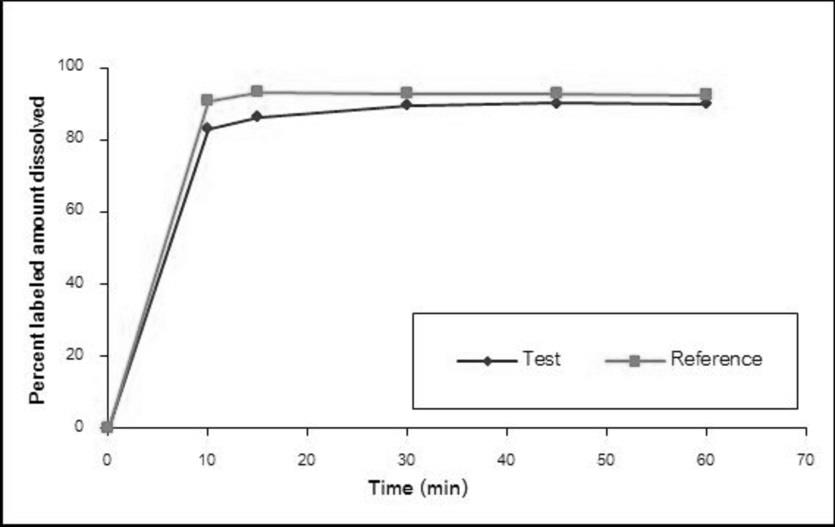
Demographic Characteristics of Subjects
Demographic characteristics of the enrolled subjects were represented in Table 1. Total 24 subjects were enrolled in the study and were randomized into TR and RT group equally. One out of 24 subjects was withdrawn due to positive urine screening test of recreational drug use during check-in of period II. Therefore, 23 subjects completed the study and their plasma concentration data were used for pharmacokinetic and statistical analysis.
| Age (year) | Weight (kg) | Height (m) | BMI (kg/m²) | |
|---|---|---|---|---|
| Mean | 36 | 60.1 | 1.65 | 22.0 |
| SD | 8.8 | 9.6 | 0.08 | 2.2 |
| Minimum | 21 | 45.8 | 1.53 | 18.4 |
| Median | 35 | 58.0 | 1.65 | 22.2 |
| Maximum | 52 | 78.8 | 1.79 | 25.4 |
Table 1: Summary of demographic data of enrolled subjects.
Sample Analysis and Incurred Sample Reanalysis (ISR)
All samples received from the clinical facility including the samples from the withdrawn subject (total 940 samples) were analyzed in 24 analytical runs. The samples from each individual were analyzed in the same analytical run. Correlation coefficient of each analytical run was more than 0.99. The accuracy and precision of bioanalysis were ensured by quality control samples in the analytical run. The coefficient of variation (CV) of quality control samples ranged from 3.9% to 5.7%. The calculated concentrations of quality control samples were between 91.7% and 94.0% of the respective nominal values.
Total 96 incurred samples were chosen for reanalysis and all of them had percent difference between the original and ISR concentrations less than 20%. The results suggested reliability and reproducibility of the analytical method (Supplementary Material).
Pharmacokinetic and Statistical Analysis
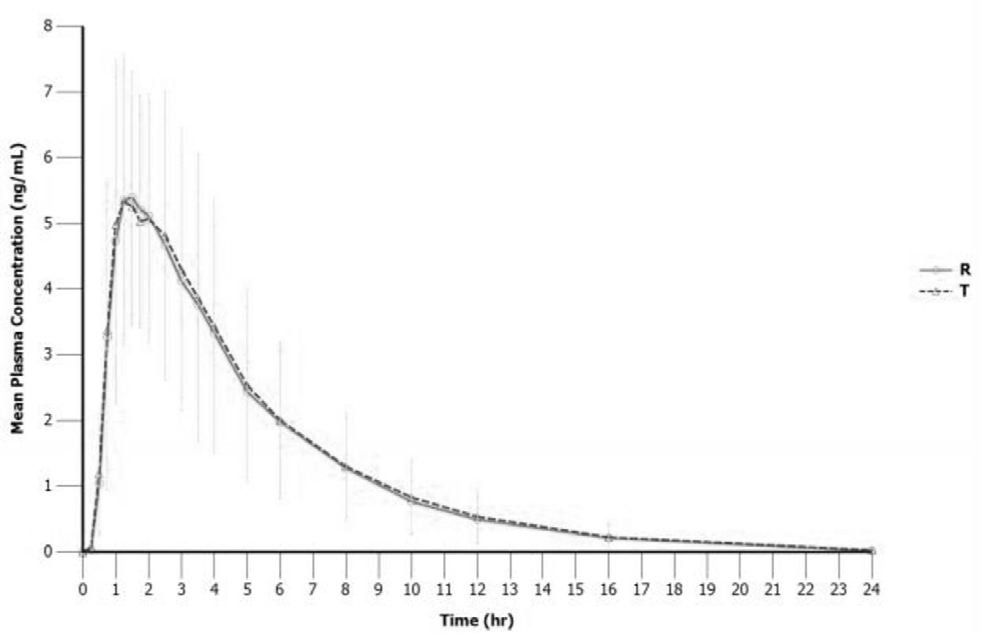
Pharmacokinetic parameters of the test and reference products are summarized in Table 2. The mean Cmax values of both products were comparable, 6.32 ng/mL for the test and 6.11 ng/mL for the reference. The mean AUC0-t values of the test and reference products were 29.6 and 28.7 hour.ng/mL, respectively and the extrapolation of AUC to infinity was less than 5%. The median tmax values were 1.25 and 1.50 hours for the test and reference products, respectively. The mean plasma concentration-time profiles of the test and reference products were relatively superimposable over sampling period (Figure 5).
| Parameter | Un-transformed data (Mean ± SD) | |
|---|---|---|
| Test | Reference | |
| AUC (hour.ng/mL) 0-t | 29.6 ± 14.9 | 28.7 ± 13.3 |
| AUC (hour.ng/mL) 0-∞ | 30.5 ± 15.1 | 29.6 ± 13.5 |
| C (ng/mL) max | 6.32 ± 2.41 | 6.11 ± 2.41 |
| t (hour)* max | 1.25 (1.00 – 3.00) | 1.50 (0.75 – 3.00) |
| λ (1/hour) Z | 0.23 ± 0.04 | 0.23 ± 0.04 |
| t 1/2 | 3.17 ± 0.55 | 3.08 ± 0.54 |
| % Extrapolated AUC | 3.20 ± 1.29 | 3.17 ± 1.32 |
Table 2: Pharmacokinetic parameters of test and reference formulations in healthy Thai volunteers.
Statistical analysis was carried out for the data derived from 23 completed subjects. The ANOVA revealed that no treatment, period and sequence effects were observed on the tested parameters, except period effect on Cmax (p < 0.05). The geometric least square mean test/reference ratios of log- transformed AUC0-t, AUC0-∞ and Cmax were 102.2%, 102.3% and 103.5%, respectively. The 90% CI of the ratios met the predefined criteria for bioequivalence. A Wilcoxon-signed rank test showed no significant difference in median tmax, p-value = 0.2348. Statistical comparisons of pharmacokinetic parameters between test and reference formulations are shown in Table 3.
| Parameters | Ratios (90% CI) | Power | Intra-subject CV (%) | ANOVA (p-value) | ||
|---|---|---|---|---|---|---|
| Treatment | Period | Sequence | ||||
| ln AUC 0-t | 102.2 (98.16-106.49) | 100.0 | 8.0 | 0.3608 | 0.0887 | 0.1528 |
| ln AUC 0-∞ | 102.3 (98.29-106.43) | 100.0 | 7.8 | 0.3406 | 0.0776 | 0.1560 |
| ln C max | 103.5 (96.91-110.51) | 100.0 | 13.0 | 0.3797 | 0.0017 | 0.4834 |
Table 3: Statistical comparisons of pharmacokinetic parameters between test and reference formulations.
Safety
Both treatments were generally well tolerated by the study subjects. No clinically relevant changes in vital sign measurements and laboratory findings were observed. The adverse event was reported for one subject with maculo- papular rash after receiving investigational product in period II. This subject was closely monitored and the adverse event could recover without medical treatment.
Discussion
A comparative in vitro dissolution study was conducted prior to the bioequivalence study to determine the possibility of establishing bioequivalence between two formulations. For the in vitro dissolution test at four different media, the dissolution profiles of the test product were found to be similar to the reference product. The same tested batches were used for in vivo bioequivalence study.
Bioequivalence study was conducted under fasting condition as the product is intended to be taken 30-45 minutes before meal [6]. Blood sampling were done more frequently during first two hours as the Cmax was anticipated between 1-2 hours after drug administration [5]. The reliability and reproducibility of the concentration data were demonstrated through precision and accuracy of the quality control samples in the analytical run, as well as the results of ISR.
Approximate 20 evaluable subjects were estimated to conduct two one-side tests for bioequivalence assuming 95% of T/R ratio, 5% significant level, 80% power, 80.00- 125.00 % bioequivalence criteria, and 21% intra-subject variability for Cmax [12]. Total 24 subjects were enrolled for a 20% overage. Therefore, only one withdrawn subject did not compromise the power of the study.
The pharmacokinetics of methylphenidate was characterized in Thai population and was in agreement with the literature data [6]. The pharmacokinetic parameters were comparable between the test and reference formulations. These findings corresponded to the results of two one- sided tests demonstrating bioequivalence between both formulations. The ANOVA model showed that period effect was statistically significant for Cmax (p < 0.05), which might arise from various sources [13]. However, in the bioanalytical phase, all plasma samples from a subject were analyzed in the same analytical run in order that a single calibration curve was used for quantification. The analysts were blinded to the randomization schedule; thus samples from two periods were treated equally. Furthermore, significant amounts of analyte were not found in any pre-dose samples collected in period II suggesting appropriate washout period and no carryover of drug administered in period I. Above all, same screening and clinical procedures were applied in both periods. The intra-subject variability for AUC0-t, AUC0-∞ and Cmax was not greater than 13.0% (Table 3). Therefore, period effect did not interfere in the results of this study.
Common adverse events of methylphenidate are insomnia, stomachache, headache, appetite reduction, and weight loss [5, 6]. Only mild adverse event was observed in this study and from the investigation, it might not be relevant to the studied drug. However, higher risk of serious adverse events and abuse potential with respect to long-term use should be considered [14]. According to the results of this study, same efficacy and safety, especially concentration- related toxicities between the test and reference products can be anticipated.
Conclusion
The pharmacokinetics of methylphenidate in Thai population was in agreement with the literature data.
This study demonstrated that Methylphenidate GPO® and Ritalin® 10, 10 mg immediate-released tablet formulations were bioequivalent with respect to their extent and rate of absorption. Based on the results, these two formulations could be used interchangeably.
Acknowledgement
This study was supported by the Government Pharmaceutical Organization (GPO), Thailand.
References
-
Tagaya H (2010) Methylphenidate: pharmacology, indication and potential of abuse. Nihon rinsho Japanese J Clin Med 68(8): 1550-1555.
-
Challman TD, Lipsky JJ (2000) Methylphenidate: Its pharmacology and uses. Mayo Clin Proc 75(7): 711-721.
-
Shier AC, Reichenbacher T, Ghuman HS, Ghuman JK (2013) Pharmacological treatment of attention deficit hyperactivity disorder in children and adolescents: clinical strategies. J Cent Nerv Syst Dis 5: 1-17.
-
Wolraich ML, Doffing MA (2004) Pharmacokinetic considerations in the treatment of attention-deficit hyperactivity disorder with methylphenidate. CNS Drugs 18(4): 243-250.
-
Morton WA, Stockton GG (2000) Methylphenidate abuse and psychiatric side effects. Prim Care Companion J Clin Psychiatry 2(5): 159-164.
-
Kimko HC, Cross JT, Abernethy DR (1999) Pharmacokinetics and clinical effectiveness of methylphenidate. Clin Pharmacokinet 37(6): 457-470.
-
Aoyama T, Kotaki H, Sasaki T, Sawada Y, Honda Y, et al. (1993) Nonlinear kinetics of threo-methylphenidate enantiomers in a patient with narcolepsy and in healthy volunteers. Eur J Clin Pharmacol 44(1): 79-84.
-
Diaz DA, Colgan ST, Langer CS, Bandi NT, Likar MD, et al. (2016) Dissolution Similarity Requirements: How Similar or Dissimilar Are the Global Regulatory Expectations? AAPS J 18(1): 15-22.
-
European Medicines Agency (2010) Guideline on the investigation of bioequivalence. Committee for Medicinal Products for Human Use (CHMP), London.
-
European Medicines Agency (2011) Guideline on bioanalytical method validation. Committee for Medicinal Products for Human Use (CHMP), London.
-
U.S. Food and Drug Administration (2018) Guidance for industry: Bioanalytical method validation. Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM), Silver Spring.
-
Schapperer E, Daumann H, Lamouche S, Thyroff- Friesinger U, Viel F, et al. (2015) Bioequivalence of Sandoz methylphenidate osmotic-controlled release tablet with Concerta® (Janssen-Cilag). Pharmacol Res Perspect 3(1): e00072.
-
Schuirmann DJ (1990) Design of bioavailability/ bioequivalence studies. Drug Inf J 24(2): 315-323.
-
Klein-Schwartz W (2002) Abuse and toxicity of methylphenidate. Curr Opin Pediatr 14(2): 219-223.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review