Bioequivalence Studies of Rivaroxaban at Two Different Strengths: Rivaroxaban 10 mg under Fasting Conditions and Rivaroxaban 20 mg under Fed Conditions in Healthy Mexican Subjects
The goal of these two studies was to evaluate the bioequivalence of two strengths of rivaroxaban 10 mg and 20 mg. Because the food effect on the pharmacokinetics of rivaroxaban affects the strength of 20 mg and not that of 10 mg. The 20 mg strength study was conducted under fed conditions, whereas the 10 mg strength study was conducted under fasting conditions. The two studies were conducted separately with different sets of Mexican subjects of both genders using a randomized, singledose, 2-sequence, 2-period cross over design with a 7-day washout period. Blood samples from the subjects were obtained at basal conditions, 0.25, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75, 2.00, 2.50, 3.00, 3.50, 4.00, 6.00, 8.00, 10.00, 12.00, 24.00, 36.00 and 48.00 hours after administration. Rivaroxaban plasma concentrations were quantified using an HPLC method coupled to a mass spectrometer. The test and reference formulations were considered to be bioequivalent if the 90% CI for the ratios (test/reference) of geometric means were within the acceptance limits of 80 to 125% for the pharmacokinetic parameters Cmax, AUC0–t and AUC0–∞. In both studies the 90% CI for all pharmacokinetic parameters were within the limits of acceptance. Therefore the conclusion of bioequivalence was reached.
Introduction
Rivaroxaban is an inhibitor of coagulation factor Xa .It is indicated in the treatment of thromboembolism, as well as in the prevention of stroke, systemic embolic events in non- valvular atrial fibrillation and major adverse cardiovascular events after acute coronary syndromes. It has an oral bioavailability >80% and a plasma half-life between 5 and 13 hours [1, 2, 3, 4]. Interestingly, there is a differential food effect on the pharmacokinetics of this drug, which depends on the drug strength. That is, the food effect affects the higher strengths (15 mg and-20 mg), while the lower strengths (2.5 mg and 10 mg) are not affected [5, 6].
Because of this effect, two separate bioequivalence studies were planned: a study with the rivaroxaban strength of 10 mg under fasting conditions and a study with the higher strength of 20 mg under fed conditions. The
sponsor of these studies, Laboratorios Liomont , S. A. de C. V. (Mexico City, Mexico), wanted to obtain the marketing approval for this drug, including these dose strengths, in Mexico. Therefore, the goal of the present research was to conduct both bioequivalence studies by comparing the two test tablet formulations containing rivaroxaban at the strengths of 10 and 20 mg with the corresponding two reference formulations: Xarelto® tablets (Bayer de Mexico, S.A. de C.V.).
Methods
Ethical Considerations
Both studies were performed according to the Declaration of Helsinki and the International Conference on Harmonisation for Good Clinical Practice Guideline. The protocols A582-20 (10-mg study) and A584-20 (20-mg study), as well as their corresponding informed consent forms were approved by the Research Ethics Committee of IPHARMA S.A. de C.V. and by the Federal Commission for Protection Against Health (COFEPRIS, Mexico). After the approval process, the studies were conducted in August (20- mg study) and in September (10-mg study) of 2021.
Subjects
For both studies, the subjects were informed and explained the potential risks and discomforts associated with the investigated drug and a written informed consent were obtained from them prior to the initiation of each study. Healthy Mexican adults of both genders (aged 18 – 55 years) were evaluated. This evaluation included a physical examination, 12-lead electrocardiogram and chest radiograph. In addition, laboratory tests (hematology, blood chemistry, urinalysis, and tests for alcohol, drug-abuse and a pregnancy test for women) and serological tests (hepatitis B and C, as well as HIV antibodies) were performed.
Formulations
For the 10 mg study, the reference formulation was rivaroxaban tablets at the strength of 10 mg (Xarelto®, Bayer de Mexico, S.A. de C.V, lot BXJEZB1, expiration date February 2022); and the test formulation as rivaroxaban tablets at the strength of 10 mg (Xaraban®, Laboratorios Liomont, S.A. de C.V., lot 337L0017-10, expiration date September 2022).
For the 20 mg study, the reference formulation was rivaroxaban tablets at the strength of 20 mg (Xarelto®, Bayer de Mexico, S.A. de C.V, lot BXJE571, expiration date April 2022); and the test formulation as rivaroxaban tablets at the strength o 20 mg (Xaraban®, Laboratorios Liomont, S.A. de C.V., lot 337L0017-20, expiration date September 2022).
Study Design
A single-blind, single-dose, randomized-sequence, two treatment, two-period crossover design with a 7-day washout interval was employed for both studies. The difference between the two studies was that the 10-mg study was designed under fasting conditions, whereas the 20-mg studies under fed conditions.
Drug Administration
For both studies, the subjects were admitted to the clinical unit (IPHARMA) and they were randomly assigned to one of the two sequences in a 1:1 ratio. The subjects were administered a single dose of the reference or the test formulation of rivaroxaban of either dose strength 10 mg or 20 mg (whichever was applicable to the corresponding study) with 250 mL of water. In the second period, the subjects received the alternative formulation, following the same procedures. For the 10-mg study, the formulations were administered after a fasting period of at least 10 hours. For the 20-mg study, the formulations were administered 30 minutes after a hypercaloric breakfast (942 kcal).
In both studies, blood samples of 6 mL were drawn from each subject. These samples were placed into heparinazed (lithium heparin) tubes. The samples were obtained at pre- dose (baseline) and then at 0.25, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75, 2.00, 2.50, 3.00, 3.50, 4.00, 6.00, 8.00, 10.00, 12.00, 24.00, 36.00 and 48.00 hours after administration. The samples were centrifuged at 3000 rpm (5°C) for 10 minutes. The resulting plasma samples were stored at -65°C ± 15°C until being transported to the analytical unit (Biokinetics) where they were maintained at -75°C ±10°C until analysis.
Determination of Rivaroxaban Plasma Concentrations
Rivaroxaban (lot: 3-JLW-16-1) and rivaroxaban-d4 (internal standard, lot: 14-VKU-116-3), were obtained from the Toronto Research Chemical (Toronto, Canada). The solvents were HPLC-mass spectrometric grade and reagents were analytical grade.
The analytical method was developed and validated by Biokinetics personnel (Mexico City, Mexico). The plasma concentrations of rivaroxaban were quantified using a HPLC method coupled to a mass spectrometer (MS/MS). A mixture of 100 μL of plasma, 10 μL of internal standard (rivaroxaban-d4, 0.08 μg/mL) and 500 μL of acetonitrile was vortexed using a 2.0 mL conical tube (Sarstedt AG & Co.) for one minute and centrifuged at 15000 rpm for 5 minutes at 20°C. The resulting supernatant was separated, transferred to a glass vial and 1µL of it was injected into the chromatographic system (ExionLC® AD System, AB Sciex, Framingham, MA).
Chromatographic Conditions
The analytical column was a Luna Omega C18 100 Å (50 × 2.1-mm internal-diameter column of 1.6-µm particle size (Phenomenex, Torrance CA). Rivaroxaban and the internal standard (IS) were eluted with a mobile phase consisting of a mixture of aqueous ammonium formate (10 mM) with formic acid (0.1%) and acetonitrile with a ratio 80:20 (v/v). The column temperature was 40°C, the flow of the mobile phase was 0.4 mL/minute, and both analytes were detected by a triple-quadrupole mass spectrometer (Sciex, model QTRAP® 6500+). The spectrometric analysis was carried out by monitoring the transition 435.991 m/z → 144.937 m/z for rivaroxaban and 439.999 m/z → 144.940 m/z for IS: The spectrometric conditions were: positive-ionization mode, declustering potential (91 V for rivaroxaban and 101 V for IS), entrance potential (10 V for rivaroxaban and 10 V for IS), collision energy (33 V for rivaroxaban and 31 V for IS), collision cell exit potential (18 V for rivaroxaban and 16 V for IS). The typical retention time for rivaroxaban and IS was 2.29 minutes. Peak areas were determined to calculate the peak area ratio of rivaroxaban with respect to that of the IS, in order to obtain the rivaroxaban concentrations.
Method Validation
The analytical method was validated in accordance with Mexican and international guidelines [7, 8]. In order to test the selectivity of the method, the validation included the use of blank human plasma samples from six different subjects, as well as hemolyzed and lipemic plasma samples, anticoagulant (lithium heparin), xanthines (caffeine and theobromine), and other drugs commonly used such as acetylsalicylic acid, diclofenac, paracetamol, ibuprofen and naproxen.
The calibration curve had the following rivaroxaban concentrations: 5, 10, 40, 160, 320, 480 and 600 ng/mL. The lower limit of quantification (LLOQ) was 5 ng/mL. The method was linear over the range of concentrations (5-600 ng/mL). In addition, sample dilution was tested to consider rivaroxaban concentrations beyond the upper bound of the range. The coefficient of determination was > 0.99 (average from 4 calibration curves). The intra-assay %CV and accuracy (relative error) of rivaroxaban were 1.80 to 3.18% and 2.08 to 8.94%, respectively; and the inter-assay %CV and accuracy were 2.42 to 4.92% and 2.25% to 7.42%, respectively. Rivaroxaban was stable in plasma for at least 25 hours at room temperature (25°C), after three freeze- thaw cycles and after 112 days at –75 ± 10°C. Quality-control samples were prepared at four different concentration levels of rivaroxaban (designated as low (15 ng/mL), medium (60 {10-mg study} and 180 ng/mL {20-mg study}) and high (450 ng/mL) independent of the calibration curve and they represented 5% of all of the tested samples.
Tolerability
The subjects were monitored during the clinical stage of both studies to determine the occurrence of adverse events (AEs). The subjects were encouraged to spontaneously report any AEs at any time over the entire duration of both studies, including washout periods.
Pharmacokinetic and Statistical Analyses
In both studies, the sample size calculations was based on the intra-subject variability of rivaroxaban Cmax with %CV of 22 for the rivaroxaban 10 mg in fasting conditions and %CV of 15 for the rivaroxaban 20 mg in fed conditions [9, 10]. The calculations, for both studies, were performed considering: 1 - β = 0.9, α = 0.05, expected ratio (μT/μR)= 0.95 and an bioequivalence acceptance limits of 80.00% to 125.00%, For the 10-mg study, the calculation yielded a minimum sample size of 30 subjects, which was increased to 34 subjects for considering potential dropouts. For the 20-mg study, the calculation yielded a minimum sample size of 16 subjects, which was increased to 22 subjects for considering potential dropouts [6, 9]. Cmax and Tmax values were obtained from the plasma concentration values. The elimination rate constant (ke) was calculated using linear regression from the terminal log-decay phase. Thus, the t½ was calculated by the quotient of ln2/ke, (where ln is the natural logarithm). The area under the curve from baseline to last quantifiable concentration (AUC0–t) was calculated using the trapezoidal method and its extrapolation from baseline to infinity was calculated as follows AUC0–∞ = AUC0–t + Ct/ke, where Ct was the last quantifiable plasma concentration. To evaluate the bioequivalence the non-compartmental method was employed and Cmax, AUC0–t and AUC0–∞ were considered to be primary variables.
The log-transformed data was analyzed using ANOVA for a 2 x 2 crossover with a significance level of 5% (α = 0.05). The 90% CIs of the geometric mean ratios (test/reference) of the Cmax, AUC0–t and AUC0–∞ were calculated. The test and the reference formulations were regarded to be bioequivalent if the 90% CIs of these parameters fell within the predetermined limits of 80.00% to 125.00%.The software package Win Nonlin Version 8.3 (Certara, Princeton, NJ, USA) was used to perform all the pharmacokinetic and statistical analyses.
Results
Table 1 reports the demographic characteristics for both studies. A total of 34 subjects were enrolled in the 10- mg study and 22 subjects in the 20-mg study. For the 10-mg study, one subject was withdrawn because he/she tested positive for drugs of abuse prior to the initiation of the study. Thus, the sample size was of 33 subjects. For the 20-mg study, none subject was withdrawn.
| Values | |
|---|---|
| Rivaroxaban 10 mg | |
| Number of enrolled subjects (female/male) | 34 (26/8) |
| Age, mean (SD), range, years | 27 (9.54), 18-53 |
| Weight, mean (SD), range, kg | 62.13 (11.99), 42.70-92.20 |
| Height, mean (SD), range, m | 1.62 (0.10), 1.48-1.87 |
| BMI , mean (SD), range, kg/m² | 23.55 (2.62), 18.72-27.00 |
| Rivaroxaban 20 mg | |
| Number of enrolled subjects (female/male) | 22 (11/11) |
| Age, mean (SD), range, years | 27 (10.22), 18-55 |
| Weight, mean (SD), range, kg | 62.61 (11.58), 43.10-91.50 |
| Height, mean (SD), range, m | 1.65 (0.11), 1.48-1.86 |
| BMI , mean (SD), range, kg/m² | 22.81 (2.56), 19.22-26.60 |
Table 1: Demographic characteristics of subjects. BMI = Body mass index SD = Standard deviation
Pharmacokinetic Parameters
Mean plasma concentration-time profiles for both studies are depicted in Figure 1. The figure suggests similar pharmacokinetic profiles for each pair of test/reference formulations corresponding to each study. Additionally, an expected delay in the time to reach Cmax (Tmax) was observed for the pair of formulations of the 20-mg study, compared to those in the 10-mg study.
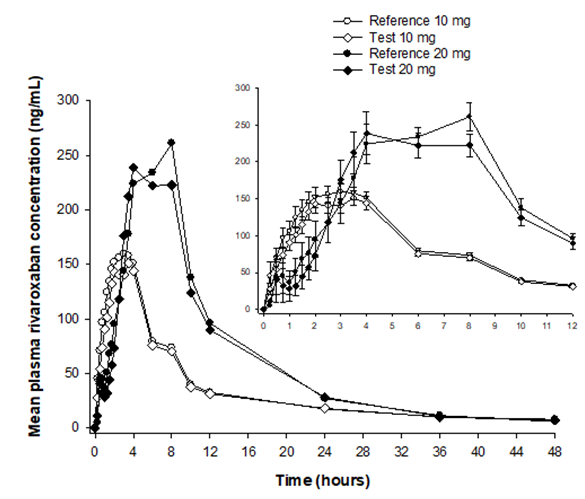
Figure 1: Mean plasma concentration-time profiles of rivaroxaban 10 mg (under fasting conditions) and 20 mg (under fed conditions). Open symbols represent the reference-test formulation pair containing rivaroxaban 10 mg (n =33). Solid symbols represent reference-test formulation pair containing rivaroxaban 20 mg (n =22). Right panel: mean (± SE) concentrations over the first 12 hours. The trademark of the reference formulations was Xarelto® (Bayer de Mexico, S.A. de C.V.) and the trademark of the test formulations was Xaraban® (Laboratorios Liomont, S.A. de C.V.)
The pharmacokinetic parameters (Cmax, Tmax, t1/2, AUC0–t and AUC0–∞) for both formulations are presented in Table 2.
| 10 mg | 20 mg | |||
|---|---|---|---|---|
| Parameter | Reference† | Test* | Reference† | Test* |
| C , ng/mL max | 195.10 (61.37) | 183.53 (61.27) | 335.25 (74.38) | 317.16 (81.10) |
| AUC , h·ng/mL 0-t | 1522.35 (339.62) | 1451.54 (376.13) | 2955.66 (593.64) | 2820.99 (635.69) |
| AUC , h·ng/mL 0-∞ | 1687.33 (381.04) | 1629.57 (395.70) | 3069.05 (593.28) | 2934.56 (626.47) |
| T , h max | 2.50[0.75, 8.00] | 2.00 [0.75, 8.00] | 6.00 [2.00, 10.00] | 4.00 [2.50, 8.00] |
| Apparent t , h 1/2 | 11.10 (4.34) | 12.86 (6.09) | 8.61 (3.02) | 9.50 (3.44) |
Table 2: Pharmacokinetic parameters of rivaroxaban after the administration of a single dose (10 mg or 20 mg) in healthy Mexican
Table 2: Pharmacokinetic parameters of rivaroxaban after the administration of a single dose (10 mg or 20 mg) in healthy Mexican subjects. Values are expressed as mean (SD) or median [range]. AUC0-t = AUC from time 0 (baseline) to the last measurable concentration AUC0-∞ = AUC from baseline extrapolated to infinity *Xaraban® (Laboratorios Liomont, S.A. de C.V., Mexico City, Mexico) †Xarelto® (Bayer de Mexico, S.A.de C.V., Mexico City, Mexico) The bioequivalence statistics for Cmax, AUC0–t and AUC0–∞: geometric means, geometric mean ratios (test/reference), 90% CI, and the intra-subject %CV is presented in Table 3.
| Parameter | Geometric means Test/Reference | Geometric Mean Ratio (%) | 90% CI | Intra-subject %CV |
|---|---|---|---|---|
| Rivaroxaban 10 mg | ||||
| C max | 173.08/185.88 | 93.11 | 83.76, 103.51 | 25.77 |
| AUC 0-t | 1405.62/1483.09 | 94.78 | 88.77, 101.19 | 15.78 |
| AUC 0-∞ | 1584.58/1641.80 | 96.51 | 90.43, 103.01 | 15.69 |
| Ribaroxaban 20 mg | ||||
| C max | 306.44/327.23 | 93.65 | 85.06, 103.11 | 18.67 |
| AUC 0-t | 2747.50/2891.41 | 95.02 | 88.21,102.37 | 14.39 |
| AUC 0-∞ | 2866.43/3008.11 | 95.29 | 88.65,102.43 | 13.97 |
Table 3: Geometric mean ratios, 90% CIs, and the intra-subject %CV of the pharmacokinetic parameter determined for rivaroxaban
Cmax = Maximum plasma drug concentration AUC0-t = AUC from time 0 (baseline) to the last measurable concentration AUC0-∞ = AUC from baseline extrapolated to infinity Table 3: Geometric mean ratios, 90% CIs, and the intra-subject %CV of the pharmacokinetic parameter determined for rivaroxaban after its single-dose administration (10 mg or 20 mg) in healthy Mexican subjects.
Tolerability
For the 10-mg study, four subjects, out of 34, reported a total of 5 AEs. These were three events of dizziness, one event of abdominal pain and one of vaginal bleeding. All of them were regarded as mild, transient and they resolved during the clinical stage. For the 20-mg study none AE was reported.
Discussion
The goal of this research was to assess the bioequivalence of two test formulations of rivaroxaban (10 mg under fasting conditions and 20 mg under fed conditions), with respect to the corresponding listed reference formulations, using two separate studies. We found for both studies, that the 90% CI of the geometric mean ratios for the 3 pharmacokinetic parameters were contained within the acceptance limits (80%-125%), indicating bioequivalence. The studied drug was well tolerated because for the 20-mg study there was none reported AE, and for the 10-mg study, there were 5 AEs reported by 4 subjects all of them of mild intensity and transient.
We acknowledge that these studies have several limitations. Like in almost any bioequivalence study, the sample size is small, making it challenging to generalize the results to large populations. Additionally, the study participants were healthy Mexican subjects within a limited range of age and body mass index, which hinders generalization to other subpopulations, such as elderly patients or those with other medical conditions. Furthermore, both studies used single doses of the drug, which prevents us from generalizing the results to its chronic use.
Conclusion
Based on the results of these studies conducted with the participation of healthy Mexican subjects of both genders, who received single doses of the rivaroxaban, we conclude that the test formulations of rivaroxaban 10 mg administered under fasting conditions, and rivaroxaban 20 mg administered under fed conditions, were found to be bioequivalent to the corresponding reference formulations, according to Mexican regulatory requirements. Additionally, these formulations were well-tolerated by the participants.
Acknowledgements
This research was supported by Laboratorios Liomont, S.A. de C.V., Mexico City, Mexico.
Conflict of Interest
The authors declare no conflicts of interest.
References
-
Gómez-Outes A, Suárez-Gea ML, Lecumberri R, Terleira- Fernández AI, Vargas-Castrillón E, et al. (2015) Direct- acting oral anticoagulants: pharmacology, indications, management, and future perspectives. Eur J Haematol 95(5): 389-404.
-
Granziera S, Hasan A, Cohen AA (2016) Direct Oral Anticoagulants and Their Use in Treatment and Secondary Prevention of Acute Symptomatic Venous Thromboembolism. Clin Appl Thromb Hemost 22(3): 209-221.
-
Abrams PJ, Emerson CR (2009) Rivaroxaban: a novel, oral, direct factor Xa inhibitor. Pharmacotherapy 29(2): 167-181.
-
Muacevic A, Adler JR, Bratsos S (2019) Pharmacokinetic Properties of Rivaroxaban in Healthy Human Subjects. Cureus 11(8): e5484.
-
Kushwah V, Arora S, Katona MT, Modhave D, Fröhlich E, et al. (2021) On Absorption Modeling and Food Effect Prediction of Rivaroxaban, a BCS II Drug Orally Administered as an Immediate-Release Tablet. Pharmaceutics 13(2): 283.
-
Zhang L, Peters G, Haskell L, Patel P, Nandy P, et al. (2017) A Cross-Study Analysis Evaluating the Effects of Food on the Pharmacokinetics of Rivaroxaban in Clinical Studies. J Clin Pharmacol 57(12): 1607-1615.
-
European Medicines Agency (2011) Guideline on bioanalytical method validation.
-
COFEPRIS (2013) Federal Commission for the Protection against Sanitary Risks. Official Mexican Standard NOM 177-SSA1-2013: Tests and procedures to prove that a medication is interchangeable [in Spanish]: General Directorate of Standards. Mexico.
-
Public Assessment Report Scientific discussion (2018) Rivaroxaban Denk 2.5 mg, 10 mg, 15 mg and 20mg, film- coated tablets rivaroxaban, NL/H/3972/001-004/DC.
-
Chow SC, Shao J, Wang H, Lokhnygina Y (2017) Sample Size Calculations in Clinical Research. In: Chow SC, et al. (Eds.), Chapman and Hall/CRC. 3rd (Edn.), Taylor & Francis group, New York, USA.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review