Generalised Severe Junctional Epidermolysis Bullosa in a Neonate-A Rare Disease
Epidermolysis bullosa is a rare group of inherited mechano-bullous disorders that manifest as blisters or erosions of the skin and in some cases the epithelial lining of other organs, in response to little or no apparent trauma. Based on ultra-structural level at which the split occurs, it is classified into simplex, junctional and dystrophic. Skin biopsy is the investigation of choice, with immunofluorescence to know the level of split. Hereby we report a case of generalized severe epidermolysis bullosa junctional in a neonate.
Abbreviations
EB: Epidermolysis Bullosa; JEB: Junctional Epidermolysis Bullosa; EBS: Epidermolysis Bullosa Simplex; DEB: Dystrophic Epidermolysis Bullosa.
Introduction
Epidermolysis bullosa (EB) is a rare group of inherited disorders that manifest as blister or erosion of the skin and epithelial lining of organs, in response to little or no apparent trauma [1]. Based on ultra-structural level at which the split occurs it can be classified into epidermolysis bullosa simplex (EBS), junctional epidermolysis bullosa (JEB) and dystrophic epidermolysis bullosa (DEB). Among these EBS is most common type. Incidence of EB as estimated by a National EB Registry report 50 cases per million live births, of these cases approximately 92% are EBS, 5% DEB, 1% JEB and 2% unclassified [2]. Herein we report a case of generalized severe JEB in a full-term newborn.
A 37-weeks term male baby, with birth weight of 2.7 kg and normal cry was born to a multigravida mother, out of non- consanguineous marriage. Patient had multiple fluid filled lesions over the hands and feet since birth which progressed to involve the whole body within 10 days. Aggravation of lesions on touching (trauma) was observed by relatives. Patient had decreased breastfeeding and developed a hoarse cry in the last 10 days.
There was history of vitiligo in father but no other autoimmune diseases in family. Elder two siblings were normal. Patient was treated with oral and topical antibiotics by private practitioner with no improvement.
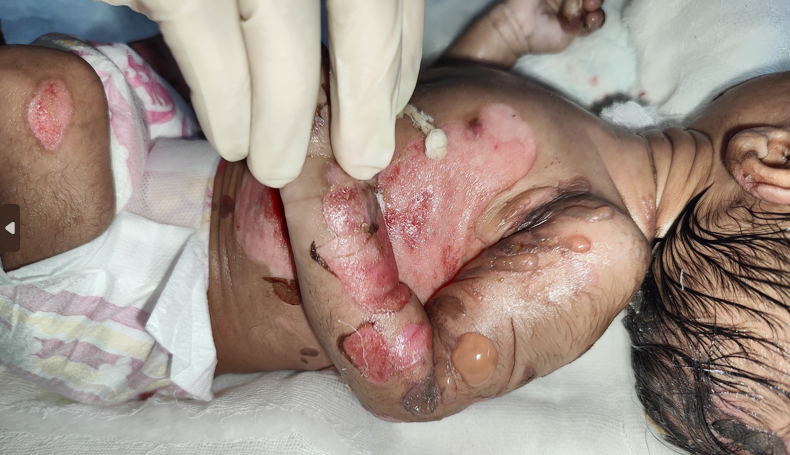
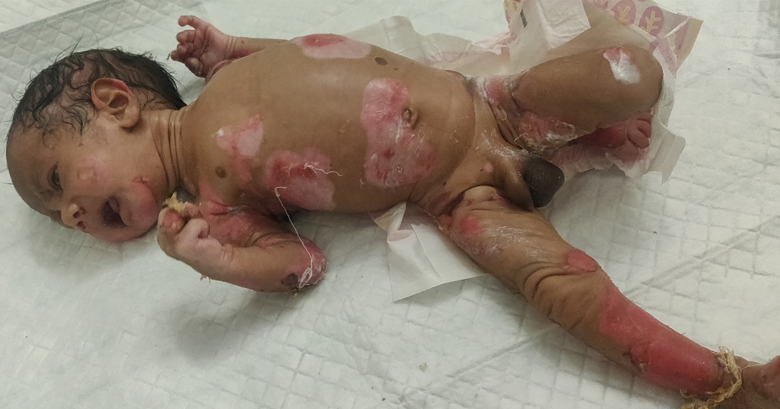
Cutaneous examination revealed multiple erosions of size approximately 2x2 to 4x5 cm over scalp, face, chest, abdomen, back and bilateral axillae, buttocks and upper and lower limbs including fingers of hands and feet but sparing palms and soles (Figures 1A & 1B). Multiple clear fluid filled bullae on an erythematous base of size 1x1 to 2x3 cm were present over bilateral upper limbs (Figure 2). A yellowish crust over scalp, fingers of bilateral hands was noted. Oral cavity showed erosions of size approximately 2x3 cm over hard palate and lips. Nails showed yellowish discoloration (Figure 3). Hair and genitals were normal. Vitals on admission were- Temperature- 36.8*C, Heart rate- 148/min, Respiratory rate- 48/min, oxygen saturation- 96% on room
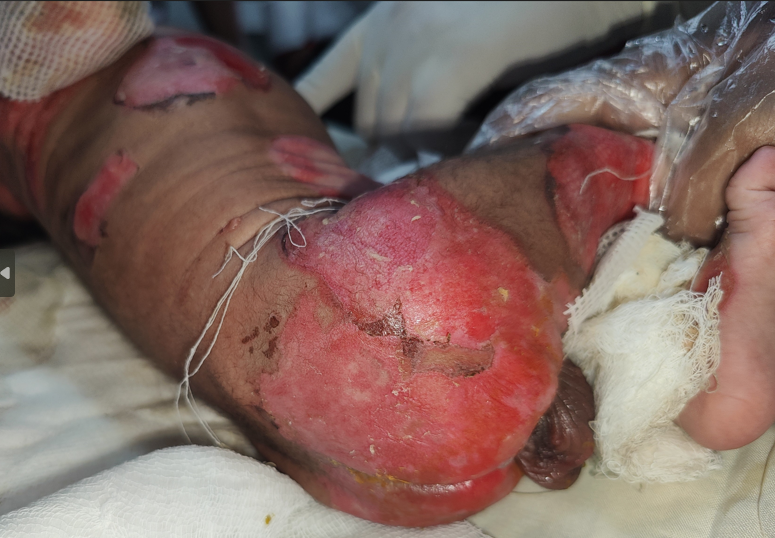
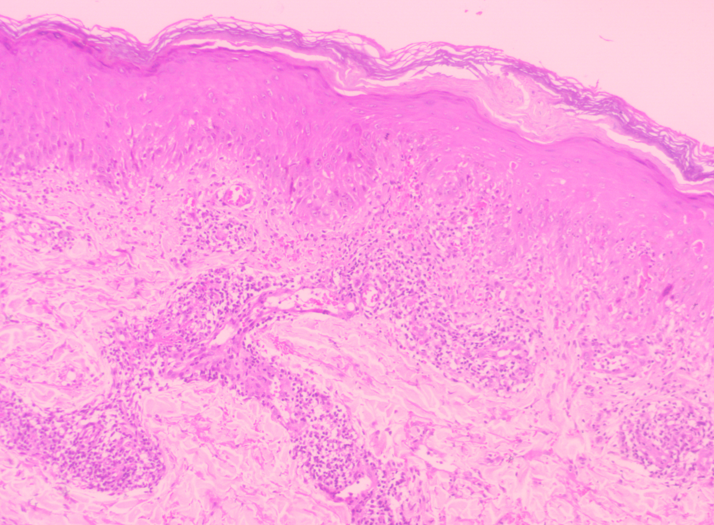
air and loud crying. Culture- Antimicrobial susceptibility testing from vesicular fluid and throat swab showed Klebsiella pneumonia, Candida Famata respectively after 48 hours of aerobic incubation. Histological examination of a skin biopsy taken from the bulla of left upper arm with differential diagnosis of EBS and JEB showed the separation located between epidermis and dermis. The blister contained neutrophilic inflammatory infiltrate with proteinaceous fluid. This confirmed diagnosis of EBJ (Figure 4).
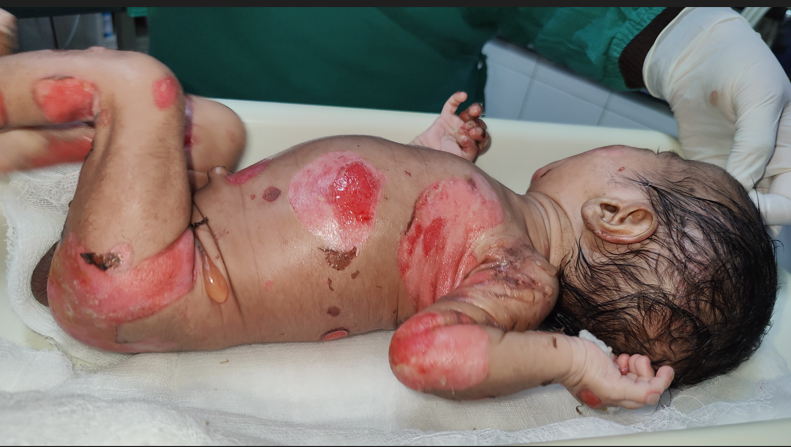
Figure 1A & B: Multiple erosions over scalp, face, chest, abdomen, back, bilateral axillae, bilateral buttocks and bilateral upper and lower limbs including fingers of hands and feet.
![Figure 4: Histology showed the separation located between epidermis and dermis. The blister contained neutrophilic inflammatory infiltrate with proteinaceous fluid. The dermis and subcutis were unremarkable. H & E stain [10X ].](/fulltextimages/13059/fig_4.png)
Patient was given syrup cotrimoxazole and paracetamol. Breast-milk was feeded with katori and spoon. Supportive management in form of daily liquid paraffin gauze skin dressings, gentamycin local application and careful handling was performed. Direct immunofluorescence and genetic testing was not done due to financial constraints. Lesions started healing with granulation tissue formation within 2 weeks (Figure 5). But infant was lost to follow up after 2 months.
Discussion
EB is broad term for a group of rare inherited mechano- bullous disorder caused by mutations in different genes. Three major phenotypes have been described depending on the level of cleavage of the basement memberane at dermoepidermal junction. The level of separation in EBS is intraepidermal, lamina densa in JEB and below basement membrane in DEB. The three phenotypes have both autosomal dominant and autosomal recessive form of inheritance [3].
JEB is autosomal recessive disorder. The prevalence is 0.3 cases per million populations and the incidence is 3.2 cases per million live births [4]. It is due to mutations in genes coding for Laminin 332, type XVII collagen, integrin a6b4 or integrin a3 causing blisters in Lamina lucida [5]. Within these subtypes, various variants like generalized severe, generalized intermediate, JEB with pyloric atresia, JEB with respiratory and renal involvement, late onset, localized, inversa and JEB- LOC (Laryngo-onycho-cutaneous) syndrome have been described. Presence of generalized skin lesions along with granulation tissue and nail changes favors JEB generalized severe type and helps to differentiate from overlapping phenotype of EB group as per clinical diagnostic matrix [6].
Various differentials in a neonate presenting with bullous lesions are autoimmune blistering diseases like pemphigus vulgaris, bullous pemphigoid and linear Ig a bullous dermatoses. Infectious etiologies like bullous impetigo, staphylococcal scalded skin syndrome and congenital syphilis should be ruled out. Mucosal involvement, presence of maternal infection, site of lesions and deposition of antibodies at various levels in skin as seen on immunofluorescence serves as basis of differentiation [7].
In EB, the histologic picture is subepidermal bulla without inflammatory cells, while the direct immunofluorescence of both lesional and non-lesional skin is negative for IgA, IgG, IgM and C3. Histopathology is generally not very helpful in delineating a specific subtype of EB, since all subtypes appear as subepidermal splits on light microscopy [5]. Skin biopsy revealed blister formation at dermo-epidermal junction with no inflammatory cells in the blister in a case report which was suggestive of JEB [8]. In our case level of split was at the dermoepidermal junction and the blister contained neutrophilic inflammatory infiltrate with proteinaceous fluid. Light microscopy also helps to exclude other infective bullous disorders. Immunofluorescence helps to diagnose immune-mediated bullous disorders showing antigen- antibody complex.
In our patient, diagnosis was based on history, assessment of clinical features and histopathology. Palms and soles were spared in our patient contrary to seen in case series of 12 patients in Kashmir valley [4] where all patients were of higher age group, as a fact that sites subjected to frequent trauma and friction are predominantly involved.
JEB since birth have limited life span owing to factors like sepsis, nutritional deficiencies and systemic complaints. Prognosis of EB depends on the severity of illness. This particular case was of generalized severe type JEB, manifesting during neonatal phase with widespread blistering and fatal progression. Patients with acral or late onset JEB have limited disease burden and can be treated symptomatically. Parents must be counseled and psychological support should be given which plays a major role in the care of patient. Supportive care to protect the skin from blistering, appropriate dressings that will not further damage the skin will aid in survival. Nutritional support is important for adequate growth and development and promotes wound healing. Genetic testing prenatally forms the basis for further family planning for a couple having child with EB diseases. Gene therapy is, potentially, a future therapy.
Conclusion
Case is reported for its rarity. All of the EB subtypes associated with various mutations have no known cure and the therapy available today focuses on pain and wound care as seen in our case. Sufficient nutrition is essential for normal growth and development as well as for wound healing. Prevention of sepsis should be the goal.
References
-
Fine JD, Eady RA, Bauer EA, Bauer JW, Tuderman L, et al. (2008) The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 58: 931-950.
-
Prasad AN (2011) Epidermolysis bullosae. Med J Armed Forces India 67(2): 165-166.
-
Qayoom S, Masood Q, Sultan J, Hassan I, Jehangir M, et al. (2010) Epidermolysis bullosa: A series of 12 patients in Kashmir valley. Indian journal of dermatology 55(3): 229-232.
-
Horn HM, Priestley GC, Eady RA, Tidman MJ (1997) The prevalence of epidermolysis bullosa in Scotland. British Journal of Dermatology 136(4): 560-564.
-
Kao CH, Chen SJ, Hwang B, Yang AH, Hsu CY, et al. (2006) Junctional epidermolysis bullosa. Journal of the Chinese Medical Association 69(10): 503-506.
-
Widhiati S, Marcella B, Dewi SR, Paramitasari AR, Ellistari EY, et al. (2019) Clinical diagnostic matrix (CDM) as a tool to diagnose subtypes of epidermolysis bullosa cases in children. Journal of General-Procedural Dermatology & Venereology Indonesia 3(2): 1-9.
-
Zhao CY, Murrell DF (2016) Blistering diseases in neonates. Current opinion in paediatrics 28(4): 500-506.
-
Srinivasan S, Sharawat IK, Saini L, Mahapatra A (2018) Junctional Epidermolysis Bullosa in a Neonate. Indian Pediatrics 55(12): 1107-1108.
- Epithelioid Granuloma; 3cases with Different Clinical Features
- Advancing Representation in Dermatology Clinical Trials: Ethical, Scientific, and Regulatory Imperatives for Inclusion Across all Fitzpatrick Skin Types
- A Case of Atopic Dermatitis with Concurrent Psoriasis Vulgaris: Successful Treatment with Upadacitinib
- Innovation Lifting Eyeshadow: A Synthesis of Makeup and Optical Illusion
- Distinguishing Superficial Actinic Porokeratosis from Actinic Keratosis with UVF Dermoscopy: A Case Report
- High Mobility Group Box 1 (HMGB1) in Cutaneous Inflammation: An Immune Modulator Bridging Cellular Stress, Ferroptosis and Danger Signaling