Fatal Case of Haemophagocytic Syndrome Associated With Infection of Plasmodium Falciparum
Haemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory condition which can occur as a primary syndrome (genetic in origin) or arise secondary to infectious, rheumatologic, malignant or metabolic disorders. HLH is characterised by hypercytokinemia and organ infiltration by phagocytozing histiocytes leading to tissue damage. This entity is often under-diagnosed, especially in adults, and specific therapy is not considered early in the disease course. We report a rare and unusual case of haemophagocytic lymphohistiocytosis (HLH) associated with infection by Plasmodium falciparum. The bone marrow aspiration and biopsy showed proliferation of histiocytes with engulfed haematopoietic cells, however patient succumbed to death before treatment for HLH could be started. Thus, this case report emphasizes the need for consideration of diagnosis of HLH in adult patients so as to provide timely diagnosis and prompt treatment to improve the clinical outcome.
Shubhangi V Agale*, Manisha P Goel, Disha D Jalan and Grace F
D’Costa
to improve the clinical outcome.
Keywords: Haemophagocytic lymphohistiocytosis; Plasmodium; Secondary
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is a potentially fatal inflammatory condition characterised by engulfment of haematopoietic cells by activated macrophages resulting in severe cytopenias [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12]. HLH can occur either as a primary syndrome (familial form) or as a secondary syndrome (acquired form). Familial HLH occurs due to an underlying genetic abnormality and has an autosomal recessive inheritance while acquired HLH is known to develop secondary to infectious, autoimmune, neoplastic or metabolic disorders. Regardless of cause, this disorder is characterized by an excessive, prolonged, and ineffective immune response. The defective cytotoxic cell function coupled with unrestrained macrophage activity leads to excessive cytokine production and tissue damage. This disorder is usually associated with high morbidity and mortality owing to its under-recognition. Immunosuppression, immune modulation, chemotherapy and haematopoietic stem cell transplant are the currently considered treatment regimens for HLH. However, early recognition is of paramount importance for therapy to prove curative in these cases [1, 2, 3]. Cases of HLH have been encountered in the past in association with viral, bacterial, fungal and parasitic infections [3, 4]. In one study conducted in tropics, infections were found to be the most common trigger for HLH among adult patients with a reported mortality of 28% [5]. Here, we report a rare and unusual fatal case of Haemophagocytic lymphohistiocytosis secondary to infection by Plasmodium falciparum.
Case Report
A 35 year old male was admitted to the hospital with chief complaints of fever since 1 week and diarrhoea, loss of weight and generalised weakness since 2 weeks. Fever was intermittent, high grade and was not associated with chills. Diarrhoea occurred on and off and was foul smelling. Patient was a chronic alcoholic. There was no significant family history. General examination revealed that the patient was febrile (101oF), dehydrated, pale and recorded blood pressure was 70/40 mm Hg. There was no lymphadenopathy, oedema or yellowish discolouration of skin and sclera. However, mild hepato- splenomegaly up to 2cm was noted. Rest of the physical examination was normal. Complete blood count on day 1 of admission revealed pancytopenia (Haemoglobin: 7.8 gm/dl; WBC count: 1620/cumm; Platelet count: 9000/cumm). Subsequently, 2 units of whole blood were transfused to the patient to improve cell counts and patient was started on antibiotics. Peripheral blood smear examination on day 1 showed the features of dimorphic anaemia along with presence of few schistocytes, hypersegmented neutrophils and pancytopenia. Platelets were normal in morphology. Reticulocyte count was less than <0.5%. In view of severe
pancytopenia, generalised weakness and chronic alcoholism, clinical diagnosis was suspected to be megaloblastic anaemia and bone marrow aspiration and biopsy were performed on third day. Peripheral smear received along with bone marrow smears showed the presence of few RBCs parasitized with ring forms of Plasmodium falciparum with a parasite index of <1% in addition to the above mentioned features. Patient was started on antimalarial artemisinin based combination therapy on 3rd day of admission. Despite initiation of anti-malarial treatment, patient continued to have fever, weakness and also developed petechiae and ecchymosis. Haematological parameters also continued to worsen. Bone marrow aspiration and biopsy were reported on 5th day of admission which revealed erythroid hyperplasia and a markedly increased number of benign looking histiocytes with small round nuclei and abundant cytoplasm, exhibiting striking phagocytosis of lymphocytes, erythrocytes and platelets. Many histiocytes also revealed the presence of engulfed pigment. However, parasite was not detected on bone marrow examination. Meanwhile, other laboratory investigations revealed hypertriglyceridemia (242mg%), increased serum ferritin (1520μg/l), increased LDH levels (637U/L) and increased serum urea (60mg%) (Table 1). Our case satisfied the guidelines of revised diagnostic criteria for HLH and diagnosis of HLH secondary to falciparum malaria was made on 6th day of admission. Patient developed sudden breathlessness for which he was intubated, however his clinical condition deteriorated rapidly and bleeding was observed in endotracheal tube. Despite of all resuscitative attempts, he finally succumbed to death due to pulmonary haemorrhage on 6th day before any immunosuppressive therapy could be instituted for treatment for HLH (Figures 1 & 2).
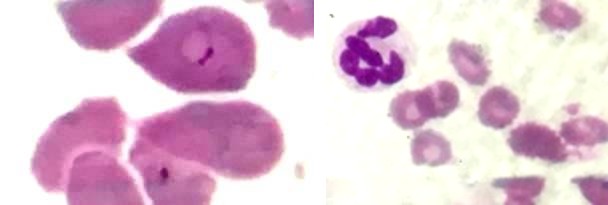
Figure 1a Figure 1b
Figures 1a & 1b: Peripheral smear showing ring forms of Plasmodium falciparum (1a), dimorphic RBCs and hypersegmented neutrophils (1b).
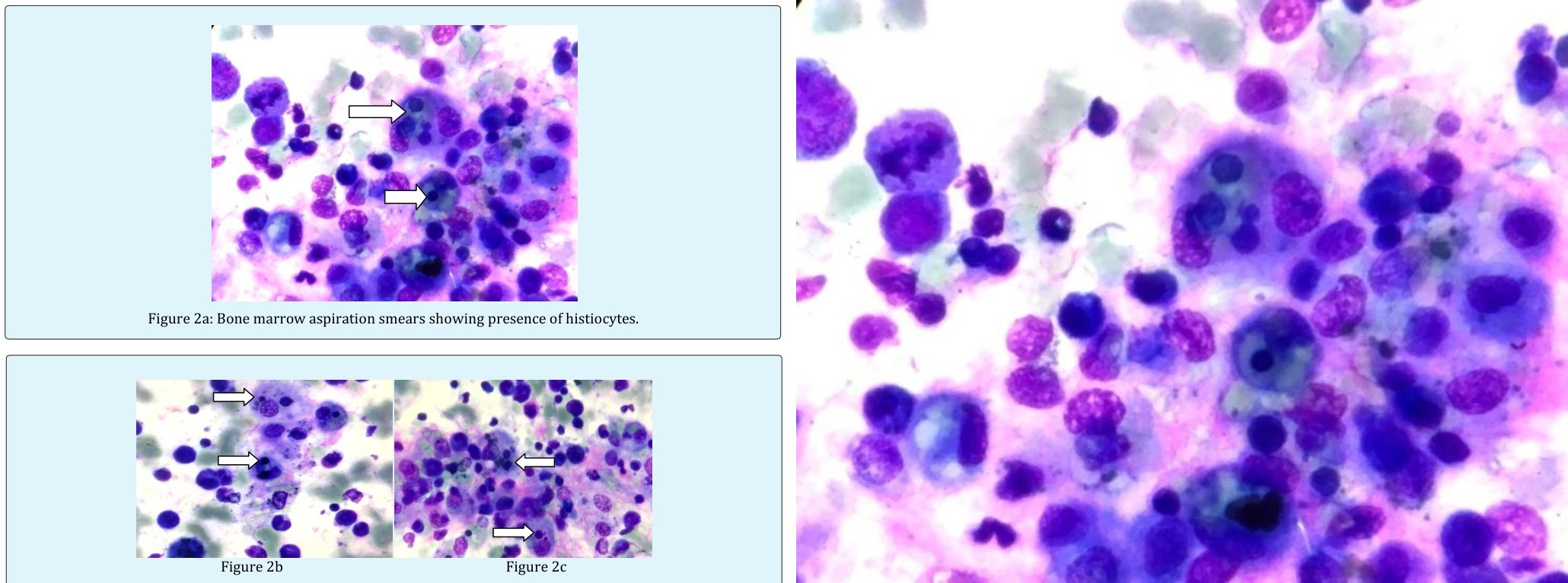
Figure 2a: Bone marrow aspiration smears showing presence of histiocytes.
Figures 2b & 2C: Bone marrow aspiration smears showing presence of histiocytes with phagocytised lymphocytes, erythroid cells and platelets (2b and 2c).
| CBC | Lipid Profile | LFT | RFT | Ferritin | Enzymes | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hb-7.8gm/dl | Triglycerides- 242mg% | SGOT- 17 U/l | Creatinine- 1.1mg% | 1520 μg/l | LDH- 637U/l | ||||||||||||
| WBC count- 1620/cumm | Cholesterol- 99mg% | SGPT- 10 U/l | Urea- 60mg% | Amylase- 246 U/l | |||||||||||||
| RBC count- 2.92million/cumm | HDL- 30mg% | Total protein- 6.6gm% | Sodium- 132Meq/L | Vitamin B12- 200ng/l | |||||||||||||
| Platelets- 9000/cumm | VLDL-48mg% | Albumin- 3.3gm% | Potassium- 4.6Meq/L | ||||||||||||||
| PCV-22.8% | LDL- 21mg% | Globulin- 3.3gm% | |||||||||||||||
| MCV-78.1 fl | Total Bilirubin- 0.6mg% | ||||||||||||||||
| MCH-26.7 pg | |||||||||||||||||
| MCHC-34.2 g/dl | |||||||||||||||||
| RDW- 18.8% |
Table 1: Investigations done in our case.
therapy is not considered early in the disease course [5]. Role of infections in causing HLH was first described in 1979 in a case series of viral associated HLH among renal transplant patients [9]. Few years later, possible bacterial etiology was discovered as well [10]. Since then, many viral (particularly EBV), bacterial, fungal and parasitic infections have commonly been implicated in causation of HLH [3, 4]. Amongst the protozoan family, Leishmania, Plasmodium primarily vivax species and Toxoplasma gondii have been encountered in cases of HLH [3, 6]. HLH secondary to Plasmodium falciparum infection was first reported by Anwar M, et al. [11] in 1995 in a young male. Literature review revealed that since then, only 20 cases of HLH secondary to malaria have been reported until 2017, highlighting the rarity of this condition [12]. Till now only twelve cases of HLH secondary to Plasmodium falciparum have been documented as shown in Table 2b [11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24]. Clinically, both familial and acquired HLH have overlapping symptomatic presentations in the form of fever, organomegaly, variable neurologic symptoms, and rarely rash, lymphadenopathy and diarrhoea.8 Laboratory findings of HLH include cytopenias, hypertriglyceridemia, hypofibrinogenemia, elevated ferritin, liver dysfunction, low or absent natural killer (NK)-cell activity, or elevated soluble CD25 (interleukin [IL]-2 receptor). Hemophagocytosis in bone marrow/spleen/ lymph nodes which is considered a hallmark of HLH may not be apparent in the bone marrow biopsy early in the disease process [3, 8]. Thus, diagnosis of HLH is based on constellation of above mentioned clinical, laboratory and histopathological features as per the guidelines by the Histiocyte Society [13]. These diagnostic criteria have been modestly modified as shown in Table 2a [8].
Discussion
HLH is a life threatening condition characterised by severe hyperinflammation due to uncontrolled proliferation of activated lymphocytes and histiocytes secreting high amounts of cytokines [1, 2, 3]. The underlying pathophysiologic mechanism of HLH is believed to be functional impairment of cytotoxic T lymphocytes and natural killer cells, resulting in failure to clear the antigenic stimulus and to turn off the inflammatory response and the ongoing tissue damage [1, 6]. The term haemophagocytosis describes the pathologic finding of activated monocytes, macrophages and histiocytes which engulf erythrocytes, leukocytes and platelets in bone marrow and other tissues and was first reported in 1939 [2]. The first case of HLH was reported by Farquhar & Claireaux [7] in 1952, as familial hemophagocytic reticulosis, a rare familial disorder characterized by a proliferation of histiocytes in solid organs and phagocytosis of blood cells [7]. HLH can be either familial (primary) or acquired (secondary). Familial forms of HLH occur due to mutations either in the perforin gene or in genes important for the exocytosis of cytotoxic granules containing perforin and granzymes. This leads to inability to induce apoptosis of target (infected) cells and ineffective down regulation of the immune response. To assist in rapid diagnosis of genetic subtypes of HLH, screening assays have been developed using intracellular staining for relevant proteins by flow cytometry [3, 6, 8]. Acquired forms of HLH are usually associated with a number of conditions. By prevalence, these include: viral infections (29%), other infections (20%), malignancies (27%), rheumatologic disorders (7%) and immune deficiency states (6%) [2, 3, 6]. However, HLH is often under-recognized, especially in adults, and specific
| 1 | Molecular diagnosis of haemophagocytic lymphohistiocytosis (HLH) or X-linked lymphoproliferative syndrome (XLP). |
|---|---|
| 2 | Or at least 3 of 4: Fever Splenomegaly Cytopenias (minimum 2 cell lines reduced) Hepatitis |
| 3 | And at least 1 of 4: Hemophagocytosis ↑ Ferritin ↑ Soluble Interleukin-2(IL-2) receptor α Absent or very decreased Natural killer cell function |
| 4 | Other results supportive of HLH diagnosis: Hypertriglyceridemia Hypofibrinogenemia Hyponatremia |
Table 2a: Modified diagnostic criteria of Haemophagocytic lymphohistiocytosis.
Studies Age (year) Gender Diagnosis of parasite Management Outcome Anwar M, et al. (1995) [11] Young adult Male Smear Antimalarials Recovered Ohno T, et al. (1996) [15] 24 Male Smear Antimalarials Recovered Retornaz, et al. (2000) [16] 73 Male Smear Sermet-Gaudelus, et al. (2000) Abdelkefi, et al. (2004) [18] 25 Male Smear Antimalarial Recovered Ohnishi, et al. (2007) [19] 30 Female Smear Antimalarials and steroids Recovered Dass, et al. (2010) [20] 16 Male Smear Antimalarials and steroids Recovered Vinoth, et al. (2011) [21] 11 months Male Bone marrow Antimalarial Recovered Rehman, et al. (2012) [22] 22 Male Smear Antimalarial Recovered Sanklecha, et al. (2012) [23] 12 Female Smear Antimalarials Recovered Nair, et al. (2013) [24] 58 Female Smear and Rapid diagnostic test (RDT) Antimalarials and IVIG Recovered Muthu V, et al. (2017) [12] 34 Female RDT Antimalarials and IVIG Recovered Present case -2018 35 Male Smear Antimalarials Death Table 2b: Association of HLH and Plasmodium falciparum reported in various studies. The above mentioned diagnostic features result due to hypercytokinemia. Fever is caused by Interleukin-1. TNF alpha leads to inhibition of lipoprotein lipase resulting in hypertriglyceridemia. Interferon-ϒ causes macrophage activation in bone marrow leading to hemophagocytosis and engulfment of heme mediated by CD 163 resulting in cytopenia and hyperferritinemia respectively. Activated lymphocytes are responsible for the production of high concentrations of soluble IL-2 receptor while hepato- splenomegaly occurs due to direct infiltration by lymphocytes and histiocytes [3]. Our patient presented with fever, hepato- splenomegaly and pancytopenia along with presence of ring forms of Plasmodium falciparum. Later on, serum ferritin and triglyceride levels were also found to be raised and the bone marrow examination revealed haemophagocytosis, thus fulfilling five criteria for diagnosis of HLH. Malarial parasite was not observed in bone marrow in our case. Even, in the previous reported 12 cases of falciparum induced HLH, only 1 case had the demonstrable parasite in bone marrow as shown in Table 2b [11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24] but still malaria was considered to be the most likely cause of HLH due to temporal relationship Antimalarials and Intravenous Immunoglobulin (IVIG) Recovered [17] 2 Male Smear Antimalarials and steroids Recovered to the illness. In our case serum LDH levels were also elevated, which, although not included in the diagnostic criteria, but have also been found to be elevated in cases of HLH reported by other studies [14]. Treatment for HLH is aimed at suppressing the hyperinflammatory state and immune dysregulation. While allogenic stem cell transplant is the only curative treatment for familial HLH, management of secondary HLH includes treatment of underlying condition along with immunosuppressive therapy in the form of corticosteroids, etoposide and cyclosporine to target the hyperactivated T cells and histiocytes as per the HLH 2004 protocol [13]. It is recommended that treatment for HLH should be started when there is a high clinical suspicion, even when results of some diagnostic studies are pending as HLH can be rapidly fatal without specific intervention [8]. In majority of the reported cases of HLH secondary to malaria, complete recovery has been observed even when the patient received the anti- malarial treatment alone, while, few required additional immunosuppression. However, no death has been reported due to falciparum malaria associated HLH as shown in Table 2b [11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24].
Our patient did not show improvement after initiation of antimalarial treatment. He subsequently developed bleeding (pulmonary haemorrhage) which could be explained by very low platelet count as a part of complication of haemophagocytosis or malarial infection. Febrile state of patient could be explained by malaria, HLH associated hypercytokinemia and possible bacteraemia due to extremely low WBC count. However, hypercytokinemia is currently believed to be the underlying factor for progressive organ dysfunction and eventually death in affected patients of HLH [8]. Hepato-splenomegaly is usually present in chronic malaria, which is endemic in India. Also chronic malaria has low parasitic index. Patient had megaloblastic anaemia and chronic alcoholism must have caused decreased immunity and damage to multiple organs. Also it is possible that he must have developed septicaemia secondary to gastrointestinal infection as chronic alcohol exposure causes damage to the wall of the intestine, allowing bacterial toxins and other harmful substances to leak from the intestine into the bloodstream. The migration of bacteria from the gut into the bloodstream also can lead to systemic infections, sepsis, and multiple organ failure [25]. Chronic alcoholics are also at increased risk of numerous complications, such as persistent fever, pneumonia, blood infections, and ARDS. Finally, they may need higher doses of certain medications to achieve effective treatment [26]. Thus cause of fatality in our case could be attributed to these various factors.
Conclusion
HLH secondary to malaria, although rare, can be a potentially fatal condition, if not diagnosed early and treated appropriately.
Recommendation
The non-specific clinical presentation of HLH can lead to diagnosis of this syndrome being delayed or overlooked. Thus, in case of persistent unresolved fever and severe cytopenias due to infections, a diagnosis of HLH should be given due consideration, as early evaluation and timely institution of immunosuppressive therapy proves imperative in such cases.
References
-
Janka GE (2007) Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr 166(2): 95-109.
-
Janka GE (2012) Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med 63: 233-46.
-
George MR (2014) Hemophagocytic lymphohistiocytosis: review of aetiologies and management. J of Blood Med 5: 69-86.
-
Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, et al. (2007) Infections associated with haemophagocytic syndrome. The Lancet infectious diseases 7(12): 814-822.
-
Rajagopala S, Singh N (2012) Diagnosing and treating hemophagocytic lymphohistiocytosis in the tropics: systematic review from the Indian subcontinent. Acta Medica Academica 41(2): 161- 174.
-
Rosado FG, Kim AS (2013) Hemophagocytic Lymphohistiocytosis: An Update on Diagnosis and Pathogenesis. Am J Clin Pathol 139(6): 713-727.
-
Farquhar JW, Claireaux AE (1952) Familial haemophagocytic reticulosis. Arch Dis Child 27(136): 519-525.
-
Filipovich AH (2009) Hemophagocytic lymphohistiocytosis (HLH) and related disorders. ASH Education Program Book 2009(1): 127-131.
-
Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour Jr HH, et al. (1979) Virus‐associated hemophagocytic syndrome: A benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 44(3): 993- 1002.
-
Risdall RJ, Brunning RD, Hernandez JI, Gordon DH (1984) Bacteria‐associated hemophagocytic syndrome. Cancer 54(12): 2968-2972.
-
Anwar M, Saleem M, Malik IA (1995) Severe haemophagocytic syndrome in falciparum malaria. J Pak Med Assoc 45(11): 302-303.
-
Muthu V, Dhooria S, Sehgal IS, Agarwal R, Behera D, et al. (2017) Malaria-associated secondary haemophagocytic lymphohistiocytosis: Report of two cases & a review of literature. Indian J Med Res 145(3): 399-404.
-
Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, et al. (2007) HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48(2): 124-131.
-
Deme S, Kakarla B, Chandra N, Subbalaxmi MV, Mallikharjuna S, et al. (2016) Hemophagocytic lymphohistiocytosis secondary to infections: a tertiary care experience. Int J Res Med Sci 4(12): 5319-5324.
-
Ohno T, Shirasaka A, Sugiyama T, Furukawa H (1996) Hemophagocytic syndrome induced by Plasmodium falciparum malaria infection. Int J Hematol 64(3-4): 263-266.
-
Retornaz F, Seux V, Arnoulet C, Durand JM, Sainty D, et al. (2000) Plasmodium falciparum malaria infection complicated by haemophagocytic syndrome in an old man. Acta Haematol 103(4): 224- 225.
-
Sermet-Gaudelus I, Abadie V, Stambouli F, Hennequin C, Lenoir G, et al. (2000) Haemophagocytic syndrome in Plasmodium falciparum malaria. Acta Paediatr 89(3): 368-369.
-
Abdelkefi A, Ben Othman T, Torjman L, Ladeb S, Lakhal A, et al. (2004) Plasmodium falciparum causing hemophagocytic syndrome after allogeneic blood stem cell transplantation. Hematol J 5(5): 449- 450.
-
Ohnishi K, Mitsui K, Komiya N, Iwasaki N, Akashi A, et al. (2007) Clinical case report: Falciparum malaria with hemophagocytic syndrome. Am J Trop Med Hyg 76(6): 1016-1018.
-
Dass R, Barman H, Duwarah SG, Choudhury V, Jain P, et al. (2010) Macrophage activation syndrome in malaria. Rheumatol Int 30(8): 1099-1101.
-
Vinoth PN, Thomas KA, Selvan SM, Suman DF, Scott JX (2011) Hemophagocytic syndrome associated with Plasmodium falciparum infection. Indian J Pathol Microbiol 54(3): 594-596.
-
Rehman JU, Bhabri N, Waleed A, Maulawi A, Aslam M (2012) Falciparum malaria in a patient with sickle cell trait with hemophagocytosis and secondary pancytopenia. Ann Hematol 91(8): 1329–1330.
-
Sanklecha M, Mehta N, Bagban H (2012) Varied presentation of complicated falciparum malaria in a family. Indian Pediatr 49(5): 413-414.
-
Nair V, Das S, Sharma A, Sharma S, Sharma P, et al. (2013) A clinicopathological analysis of 26 patients with infection-associated haemophagocytic lymphohistiocytosis and the importance of bone marrow phagocytosis for the early initiation of immunomodulatory treatment. Postgrad Med J 89(1050): 185-192.
-
Hammer AM, Morris NL, Earley ZM, Choudhry MA (2015) The first line of defense: The effects of alcohol on post-burn intestinal barrier, immune cells, and microbiome. Alcohol Res 37(2): 209-222.
-
Trevejo-Nunez G, Kolls JK, de Wit M (2015) Alcohol use as a risk factor in infections and healing: A clinician’s perspective. Alcohol Res 37(2): 177-184.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies