Incidental Detection of Sickle Cell Anaemia as a Significant Haematological Problem in an Island Based Population
Introduction: Sickle cell anaemia is a haematological disease with a known gentic cause. However, there is paucity of data regarding this condition from islands of the world. Also there is no previous study in which incidentally sickle cell anaemia has been detected at a mass scale in a large number of cases as part of celebration of a national event. Materials & Methods: The study was carried out on 500 students who were participating in the republic day celebrations on 26th January 2017 at Kavaratti island, Lakshadweep. Amongst these students 250 complained of drowsiness and pain in leg, knees and chest. They were given immediate medical attention and blood samples were drawn. In the laboratory complete blood count, peripheral smear examination and sickling test were performed. The haematological data was analyzed and correlated. Results: Out of total 250 cases, 150 (60%) showed positive sickle cell test. Peripheral smear examination showed that in 65 out of these 150 cases (43.3%) there was a picture of Sickle thalassemia (HbSThal). In the rest of 85 cases (57%) the smears showed variable presence of sickle cells ranging from occaisional to many. The haemoglobin in all these cases ranged from 10-14.5 g/dl Conclusion: Present study is the first report of its kind in which sickle cell anaemia has been detected incidentally in a large number of cases simultaneously as part of celebration of a national event. It also highlights importance of fluid replacement therapy in cases of sickle cell anaemia who develop vaso-occlusive crises.
Introduction
Sickle - cell disease or sickle cell anaemia (or depanocytosis, HbS) is a haematological disorder with a genetic basis. It is characterized by red blood cells that conditions of low oxygen assume an abnormal, rigid, sickle shape. The condition was unknown until the discovery of the sickle cells in1904 by the Chicago based cardiologist and a professor of medicine James B. Herrick (1861 - 1954) whose intern Ernest Edward Irons (1877 - 1959) observed" peculiar elongated and sickle - shaped" cells in the blood of Walter Clement Noel, a 20 - year - old first year dental student from Canada after Noel was admitted to the Chicago Presbyterian Hospital in December 1904 suffering from anaemia [1]. Noel was readmitted several times over the next three years for "Muscular rheumatism" and "bilious attack". Vernon Mason named the disease as "Sickle cell anaemia" in 1922. Linus Pauling and his colleagues for the first time, in 1949, demonstrated that sickle cell disease occurs as a result of an abnormality in the haemoglobin molecule. This was the first time a genetic disease was linked to a mutation of a specific protein, a milestone in the history of molecular biology [2]. In India sickle hemoglobin was first discovered by Lehmann & Cutbush [3] more than 50 years ago among the tribal's of Nilgiri Hills of Southern India [3]. Subsequent studies conducted by various workers have reported high frequencies throughout Central India and parts of Southern India. The present prevalence rate among tribal people varies from 10-25% in certain regions [4, 5]. The process of sickling is initially reversible but later on becomes irreversible. Sickling decreases the cells' flexibility and may lead to a variety of complications. The sickling occurs because of a mutation in the haemoglobin gene. In this there is a point mutation in the β-globin chain of haemoglobin at the 6th position glutamic acid is replaced by valine. This occurs because of a nucleotide change, adenine to thymine (GAG→GTG) of codon 6 of β- globin gene. The overall effect of this mutation is that it alters the net charge of hemoglobin, oxygen affinity and renders it unstable. Under deoxygenated conditions, the solubility of HbS decreases whereas the viscosity is increased. There is formation of a gel-like substance containing Hb crystals called tactoids [6, 7]. The exposure of RBCs that contain the gel, to deoxygenated state initiates the process of sickling. Sickled hemoglobin gets polymerized and deforms the red blood cell from discoid shape to sickle shape. Sickle cell anaemia is transmitted as an autosomal recessive disorder. That is if a person receives only one gene responsible for sickle hemoglobin from either of the parent, the condition is called carrier or trait. However, if one inherits two defective genes, one from each parent, the condition is called Sickle cell anaemia. Individuals with trait lead a normal life but the diseased person suffers from various complications throughout the life, such as anemia, bone & joint pain, joint swelling, recurrent infection, osteomyelitis, necrosis of bone, aplastic crises, abdominal pain, Splenic sequestration crises, hepatosplenomegaly etc. Sickle cell anaemia usually manifests early in childhood. For the first 6 months of life, infants are protected largely by elevated levels of HbF. Sickle cell anaemia is believed to provide protective benefit against Malaria infection. That is why the prevalence of Sickle cell anaemia is far higher in areas that are endemic for Malaria. The mechanism for this protective effect is as follows. The presence of malaria parasite causes the red blood cells with defective haemoglobin to rupture prematurely making the plasmodium unable to reproduce. Furthermore, the polymerization of Hb affects the ability of the parasite to digest Hb in the first place. Therefore, in areas where malaria is a problem, people's chances of survival improve if they carry sickle cell trait. There is paucity of literature on Sickle cell anaemia from the remote islands of the world. Most of the published literature is on the study of clinical, haematological and molecular parameters in these patients. However, to the best of our knowledge there is no published report of incidental mass detection of Sickle cell anaemia in an island based population.
Materials & Methods
The study was carried out on 500 students who were participating in the republic day celebrations on 26th January 2017 at Kavaratti island, Lakshadweep. These students were marching in a hot and humid environment. Almost half of these students (250) complained of drowsiness and pain in leg, knees and chest. They were immediately taken to the mobile emergency medical van stationed at the venue. After clinical examination appropriate immediate medical management was done.
Blood samples of all these cases were withdrawn and sent to the laboratory of Indira Gandhi Hospital, Kavaratti. In the laboratory complete blood count, peripheral smear examination and sickling test (where ever necessary was performed in all the samples. The haematological analysis was carried out on a MEK-6420 haematology analyser (Nihon-Kohden). The haematological data was correlated with other ancillary variables wherever relevant. Statistical analysis using appropriate tools was done.
ranged from 13 to 17 years while amongst females it was 12 to 18 years. In clinical history none of the patients had a history of painful limbs in the last one year, hand foot syndrome, history of hospitalization or blood transfusion. On physical examination all of these students were found to be having a weak, thready pulse. The pulse rate ranged from 96-114. The systolic blood pressure was from 90-110 mm Hg and diastolic blood pressure varied from 50-70 mm Hg. None of these students were obese and had any other significant past or family medical history. The features of dactylitis were not seen in any of the cases. Hepatomegaly (<4 cm) was seen in 3 cases. However, in none of the cases it was > 4 cm. Splenomegaly was not seen in any of the cases. The results of theses clinical parameters are summarily presented in Table 1.
Results
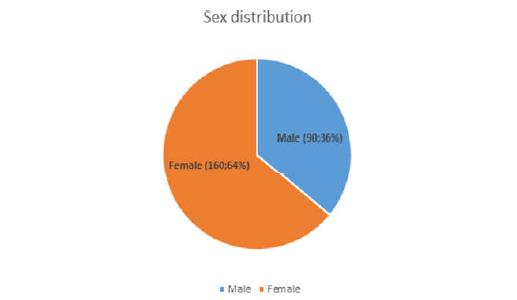
The study included a total of 250 students. These included 90 males (36%) and 160 females (64%) (Figure 1). The M: F ratio was 1: 1.8. Amongst the males age
| History/Symptom | Present | Absent | ||||||
|---|---|---|---|---|---|---|---|---|
| Painful limbs in the last one year | 0 | 250 | ||||||
| Hand foot syndrome | 0 | 250 | ||||||
| History of hospitalization | 0 | 250 | ||||||
| Blood transfusion | 0 | 250 | ||||||
| Physical examination | ||||||||
| Hepatomegaly (< 4 cm) | 3 | 247 | ||||||
| Hepatomegaly (> 4 cm) | 0 | |||||||
| Splenomegaly | 0 | 250 | ||||||
| Dactylitis | 0 | 250 |
Table 1: Clinical parameters in the 250 cases. They were infused normal saline and 50% dextrose saline. Out of the 250 students 9
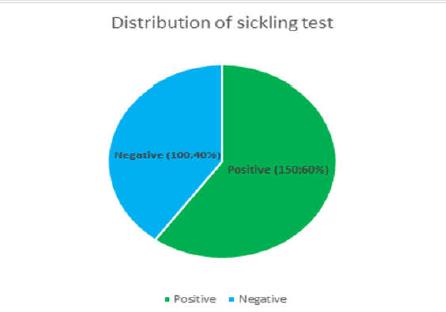
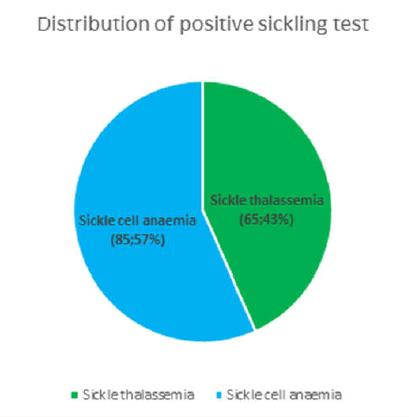
Table 1: Clinical parameters in the 250 cases. They were infused normal saline and 50% dextrose saline. Out of the 250 students 95 required non-narcotic analgesics for relief. The students recovered well with medical management and rejoined the event after an interval of 15-40 minutes. Blood samples of all these cases were withdrawn and sent to the laboratory of Indira Gandhi Hospital, Kavaratti. In the laboratory complete blood count, peripheral smear examination and sickling test was performed in all the samples. Out of all the cases 150 (60%) showed positive sickle cell test whereas the rest of 100 cases (40%) were negative (Figure 2). Peripheral smear examination showed that in 65 out of 150 cases (43.3%) there was a picture of Sickle thalassemia (HbSThal). The smears in these cases showed sickle cells and target cells (Figure 3). The RBC indices were microcytic hypochromic. In the rest of 85 cases (57%) the smears showed variable presence of sickle cells ranging from occasional to many (Figure 4). These cases were labelled as sickle cell anaemia and were advised further workup by HPLC. The overall haemoglobin level in these 150 cases varied from 10-14.5 g/dl. The distribution of these 150 cases which showed positive sickeling test is shown in Figure 5.
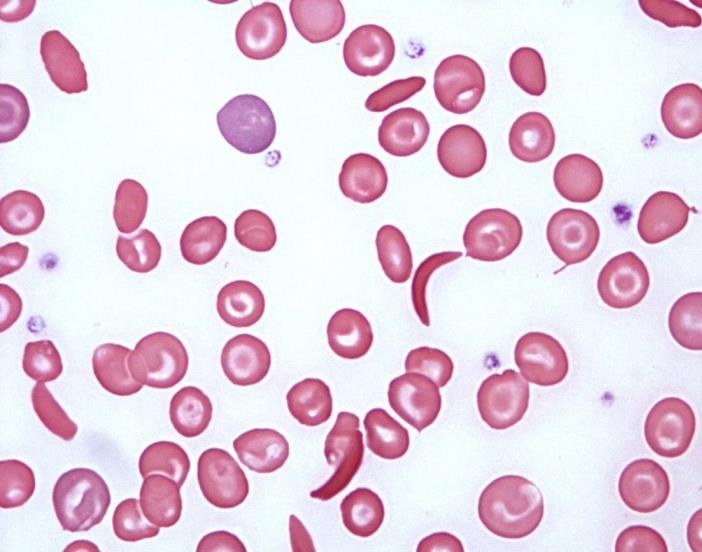
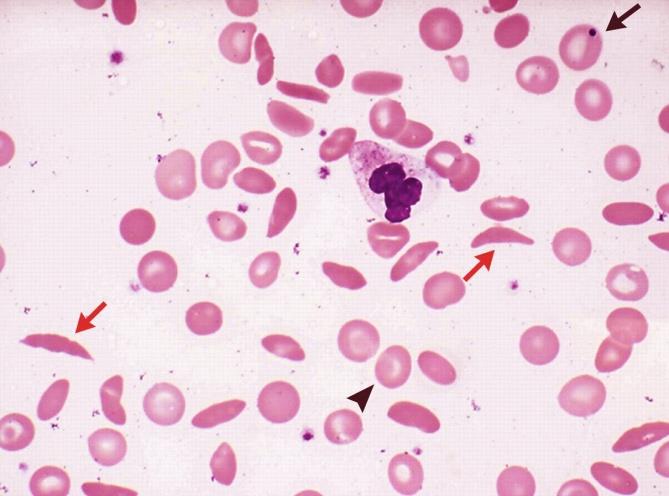
Sickle cell anaemia (SCA) is an inherited condition of chronic haemolytic anaemia that includes many genotypes. Amongst the important complications of Sickle cell anaemia are vaso-occlusive crises, haemolytic crises [8, 9, 10, 11, 12], sequestration crises [13] and immune deficit [14, 15, 16, 17, 18, 19]. Vaso-occlusive crisis (VOC) involving the back, legs, knees, arms and abdomen is amongst the clinically very important complication which is known to occur in sickle cell anaemia. It is the commonest reason for hospital visit in these patients [20]. Recurrent episodes of these crises lead to complications such as stroke, nephropathy, a vascular necrosis of bone, pulmonary hypertension and retinopathy. VOC in SCA also leads to injury to multiple organ systems. It is well known that large numbers of sickled red cells lodging in the microvasculature appear not to be the only link to causation of vaso-occlusion rather, other factors play significant roles such as excessive tendency of both sickle and non sickled red cells to adhere to vascular endothelium and subsequent activation of other cell lines like platelets and leukocytes [21, 22]. These crises get precipitated by dehydration which in this case occurred due to exposure to sunlight and stress.
promoting sickling of red cells and thereby facilitates the occurrence of painful crises. A relationship between dietary intake of sodium and water in patients of SCA has been seen. It has been observed that in these patients water intake is significantly lower than what is adequate. Due to this low water intake the concentration of sodium rises. The situation gets further compounded by the high temperature and humidity at Kavaratti, Lakshadweep which leads to profuse sweating and further loss of water from the body. An additional benefit of hydration was noted in a study by Okomo, et al. [23] who found that when sickle cell patients increase fluid intake the occurrence of vaso- occlusive crises was significantly decreased. Although SCA patients remain asymptomatic usually but under conditions of dehydration and stress vaso-occlusive crises manifest themselves. Since these students are the future of any nation it is crucial to take good care to bring out the best from them. Haemoglobin was found to be quite near to the normal range in these students. This can be attributed to the extensive consumption of fish in the local diet which is known to be an excellent source of haem iron. Thus children from coastal areas of India like Kerala, Maharashtra, Tamil Nadu, Andhra Pradesh, Andaman &Nicobar islands, Lakshadweep islands should be treated with extreme care when they participate in any national or international event which involves prolonged stress and exposure to hot and humid weather. There is extremely high prevalence of haemolyticanaemia specially sickle cell anaemia in Lakshadweep. These individuals are thus at a much higher risk of developing complications like vaso- occlusive crises. There is a need for counselling of students from Lakshadweep who wish to participate in strenuous physical exercise and sports events at national, international and even school level. However, the overall life span and quality of life can be absolutely normal if appropriate fluid management is instituted at the right time. The present study is the first report of its kind in which sickle cell anaemia has been detected incidentally in a large number of cases simultaneously as part of celebration of a national event.
References
-
Herrick JB (1910) Peculiar elongated and sickle shaped red blood corpuscles in a case of severe anemia. Arch Intern Med 6: 517.
-
Pauling L, Itano H, Singer SJ, Wells IC (1949) Sickle cellanemia: A molecular disease. Science 110(2865): 543-548.
-
Lehmann H, Cutbush M (1952) Sickle-cell trait in southern India. Br Med J 1(4755): 404-405.
-
Bhatia HM, Rao VR (1987) Genetic Atlas of the Indian tribes institute of immunohematology, Indian council of medical research (ICMR), Bombay, India.
-
Kar BC (1991) Sickle cell disease in India. J Assoc Phys India 39: 954-960.
-
Edelstin SJ (1981) Molecular Topology in Crystals and Fibres of Haemoglobin S. J MoL Biol 150(4): 557- 575.
-
Kwiatkowski DP (2005) How Malaria has affected the Human Genome and what Human Genetics can teach us About Malaria. Am J Hum Genet 77(2): 171- 192.
-
Kaul DK, Fabry ME, Nagel RL (1986) Vaso – Occlusion by Sickle Cells: Evidence for Selective Trapping of Dense red Cells. Blood 68(5): 1162- 1166.
-
Crosby WH (1955) The Metabolism of Haemoglobin and Bile Pigment in Haemolytic Disease. Am J Med 18(1): 112-122.
-
Allan D, Limbrick AR, Thomas P, Westerman MP (1982) Release of Spectrin - Free Spicules on Reoxygenation of Sickled Erythrocytes. Nature 295: 612-613.
-
Padlia F, Bromberg PA, Jensen WA (1973) The Sickle- Unsickle Cycle: A Causes of Cell Fragmentation Leading to Permanently Deformed Cells. Blood 41: 653-660.
-
Test ST, Kleman K, Lubin B (1991) Characterization of the Complement Sensitivity of Density - Fractionated Sickle Cells. Blood, Suppl 78: 202.
-
Winkelstein JA, Drachman RH (1966) Deficiency of Pneumococcal Serum Opsonizing Activity in Sickle Cell Disease. N Eng J Med 279(9): 459-466.
-
Hosea SW, Burch CG, Brown EJ, Berg RA, Frank MM (1981) Impaired Immune Response of Splenectomized Patients to Polyvalent Pneumococcal Vaccine. Lancet 1(8224): 804-807.
-
Pearson HA, Gallagher R, Chilcote E, Sullivan E, Wilimas J (1985) Development Pattern of Splenic Dysfunction in Sickle Cell Disorder. Paediatrics 76(3): 392-397.
-
De Ceuler K, Wilson WA, Morgan AG, Serjseant GR (1981) Haemoglobinemia and Activation of the Complement System in Homozygous Sickle Cell Disease. J Clin Lab Immunol 6: 57.
-
Carr DJ, Guarcello V, Blalock JE (1992) Phosphatidylserine Suppresses Antigen-Specific IgM Production by Mice Orally Administered Sheep Red Blood Cells. Proc Soc Exp Biol Med 200(4): 548-554.
-
Franek PF, Bevers EM, Lubin BH (1985) Uncoupling of the Membrane Skeleton from the Lipid Bilayer.The Cause of Accelerated Phospholipids Flip -Flop Leading to an Enhanced ProcoagulantActivity of Sickled Cells. J Clin Invest 75: 183.
-
Gaston MH, Verier JI, Woods G, Pegelow C, Kelleher J (1986) Prophylaxis with Oral Penicillin in Children with Sickle Cell Anaemia. N Engl J Med 314(25): 1593-1599.
-
Ahmed AE, Alaskar AS, McClish DK, Ali YZ, Mohammed H. Aldughither, et al. (2016) Saudi SCD patients’ symptoms and quality of life relative to the number of ED visits. BMC Emergency Medicine 16(1): 30.
-
Akinbami A, Dosunmu A, Adediran A, Oshinaike O, Adebola P, et al. (2012) Haematological values in homozygous sickle cell disease in steady state and haemoglobin phenotypes AA controls in Lagos, Nigeria. BMC Research Notes 5: 396.
-
Ballas SK, Lieff S, Benjamin LJ, Dampier CD, Heeney MM, Hoppe C, et al. (2010) Definitions of the phenotypic manifestations of sickle cell disease. Am J Hematol 85(1): 6-13.
-
Okomo U, Meremikwu MM (2017) Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane Database Syst Rev 7: CD005406.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies