The Stellar, Astral Reproduction: Burkitt`S Lymphoma
An infrequent (≤1.2%), aggressive B lymphocyte non hodgkin’s lymphoma is designated as a Burkitt’s lymphoma, which may present as an extra-nodal tumefaction or as an acute leukaemia [1]. A well delineated clinico-pathological disorder, Burkitt’s lymphoma elucidates minimal morphological, phenotypic and molecular diversity. The proliferating tumour displays a rapid doubling time of approximately twenty five hours. The clinical sub-categories of the Burkitt’s lymphoma may be: i) endemic Burkitt’s lymphoma (e BL) ii) sporadic Burkitt’s lymphoma (s BL) and iii) immune deficiency related Burkitt’s lymphoma (id BL) [1]. A frequent, paediatric class of Burkitt’s lymphoma (4-12 years) with an extra-nodal representation of the jaw or assorted facial bones(50-60%), is the ENDEMIC variant with an identical geographic apportionment as the Plasmodium falciparum. The primary tumour may disseminate to the mesentery, ovary, testis, breast and meninges. Limited to the African children, the endemic variant may account for roughly half (30-50%) of the childhood malignancies. An estimated 3-6 patients /100000 population per year may be detected
Preface
An infrequent (≤1.2%), aggressive B lymphocyte non hodgkin’s lymphoma is designated as a Burkitt’s lymphoma, which may present as an extra-nodal tumefaction or as an acute leukaemia [1]. A well delineated clinico-pathological disorder, Burkitt’s lymphoma elucidates minimal morphological, phenotypic and molecular diversity. The proliferating tumour displays a rapid doubling time of approximately twenty five hours. The clinical sub-categories of the Burkitt’s lymphoma may be: i) endemic Burkitt’s lymphoma (e BL) ii) sporadic Burkitt’s lymphoma (s BL) and iii) immune deficiency related Burkitt’s lymphoma (id BL) [1]. A frequent, paediatric class of Burkitt’s lymphoma (4-12 years) with an extra-nodal representation of the jaw or assorted facial bones(50-60%), is the ENDEMIC variant with an identical geographic apportionment as the Plasmodium falciparum. The primary tumour may disseminate to the mesentery, ovary, testis, breast and meninges. Limited to the African children, the endemic variant may account for roughly half (30-50%) of the childhood malignancies [1, 2]. An estimated 3-6 patients /100000 population per year may be detected [3].
Extra-nodal, abdominal sites (91%) of the children and young adults such as the terminal ileum, cecum, intra- abdominal lymph nodes, stomach, appendix, kidney, ovary, breast, mesentery, testis bone marrow (20%), central nervous system with massive disease and ascites may frequently appear in the SPORADIC sub-type. The infrequent bone marrow involvement may enhance in the adults. Symptoms of bowel obstruction, gastro-intestinal bleed, acute appendicitis, intussusceptions and localized lymph node enlargement may appear [1, 2]. Immune- Deficiency associated Burkitt’s lymphoma may commonly arise secondary to auto-immune disease or in HIV positive individuals elucidating the primary or acquired auto-immune deficiency syndrome (AIDS) or infrequently with immune-modulation therapy following a transplant or in patients with primary immune disorders (PIDs) [1]. The extra-nodal disease may exceed the nodal emergence. The aggressive sub-type may appear as an oncologic emergency and is predominantly chemo-sensitive. The initial incidence was roughly one third (30-40%) of the lymphomas and is currently declining following the employment of highly active anti-retroviral therapy (HAART). Burkitt’s lymphoma may involve individuals with elevated CD4+ counts (> 200µl) with an absence of opportunistic infections. The lymphoma may not proportionately recede in the infected (HIV+) population with the onset of highly active antiretroviral therapy (HAART), in contrast to the HIV associated lymphomas. Immune deficiency variant may implicate the lymph node and display symptoms of central nervous system involvement. Extensive blood and bone marrow dispersal with Burkitt’s leukaemia may exemplify [2, 3].
The immune suppression associated Burkitt’s lymphoma may depict marked cytological variations. Morphological differentiation between Burkitt’s lymphoma and high grade B cell lymphoma not otherwise specified (HG BCL NOS), Burkitt’s like lymphoma with 11q aberration and diffuse large B cell lymphoma (DLBCL) with aggressive aspects may be challenging. A comprehensive immune phenotypic and fluouroscent in situ hybridization (FISH) evaluation may be a pre- requisite [2, 3, 4].
Histo-Morphology
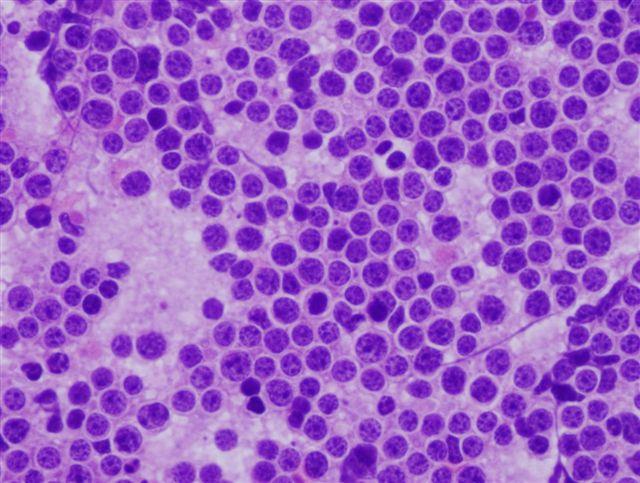
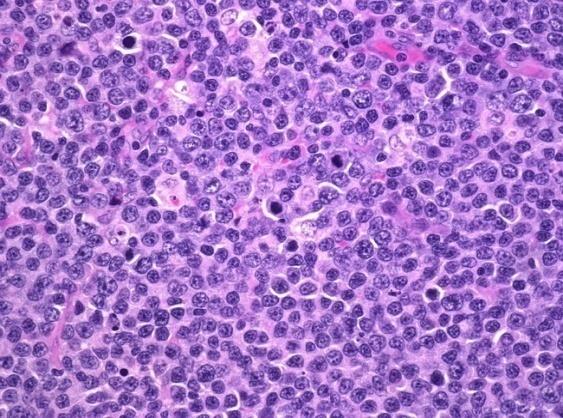
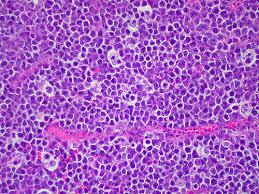
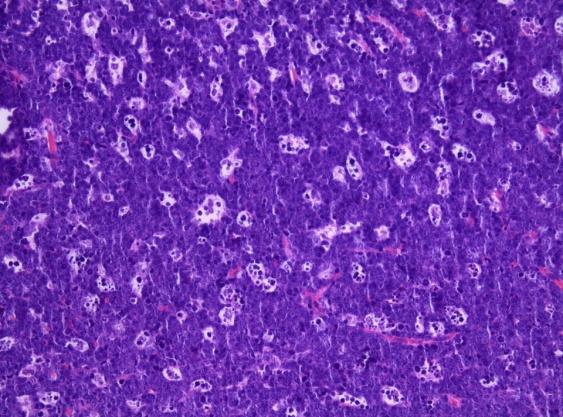
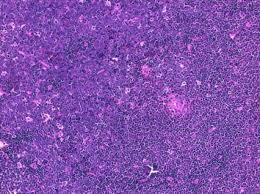
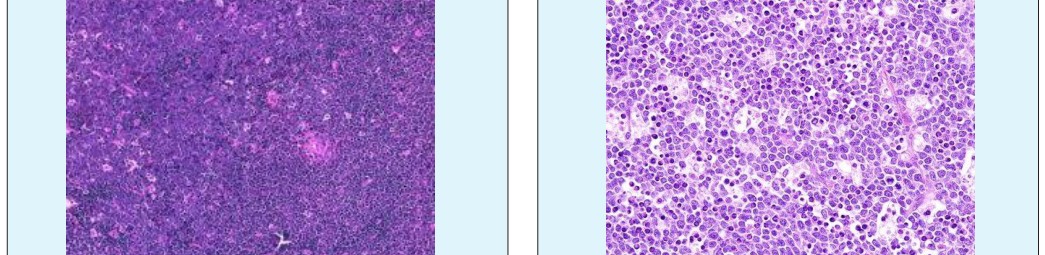
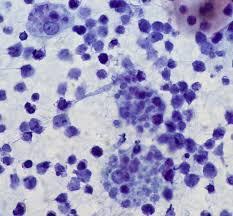
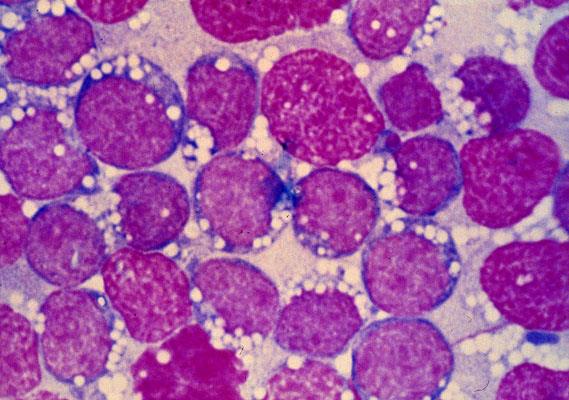
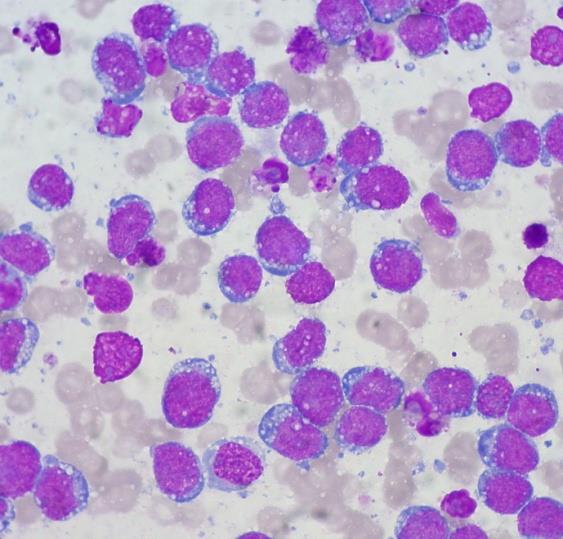
A surgical excision biopsy may be opted for instead of a fine needle aspiration (FNAC) or a core needle biopsy for the histological elucidation of lymphoid proliferations including a Burkitt’s lymphoma. A fine needle cytology or a core biopsy may be beneficial in a problematic approach to a lesion or a re-occurring nodule. Classically composed of uniform, medium sized lymphocytes with numerous, miniature round nucleoli, finely dispersed chromatin and scanty, basophilic vacuolated cytoplasm , the monotonous cellular configuration may depict a cohesive dispersal with a frequent “ starry sky “ arrangement by virtue of the multiple tingible body macrophages. Mitosis may be numerous along with fact vacuoles in the cytoplasm. The “plasmacytoid “variant depicts immunoglobulin containing eccentric basophilic cytoplasm with a single, prominent nucleolus. The “atypical” or the “ pleomorphic “ variant comprises of large cells with a definite pleomorphism [5].
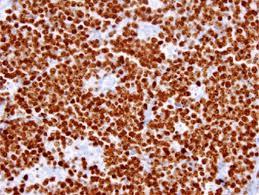
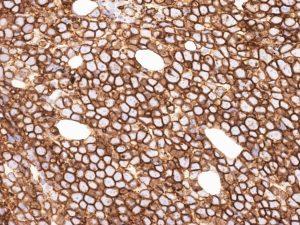
The ultra-structure depicts abundant ribosomes, lipid inclusions, an absence of glycogen articles and nuclear projections [5]. The typical immune phenotype elaborated by the lymphoma cells may be : CD10+, CD20+, CD79a+, Ki67+ (MIB 1 100%), BCL6+ CD38+, Ig M, CD5-( rare T cells), Tdt -, BCL2-, MUM1/IRF4-, CD44-, CyclinD1-, TP53 and c MYC +(80%). PAX 5+, CD43+, plasma cell antigen CD38+. The Epstein barr encoded RNAs (EBERs) may be delineated maximally in the endemic variant (100%) followed by the immune-deficient type(40%) and least in the sporadic category(30%)(1,3). The viral latency may be characterized by the presence of Epstein barr nuclear antigen (EBNA1+) and the lack of latency membrane antigen (LMP1-). The characteristic cytogenetic aberration may be the over-expression of c MYC onco- gene on account of the t(8:14)(q24:q32) or uncommonly a t(8:22)(q24:q11) or a t(2:8)(q24:p12) translocation. The aberration may be discerned in the majority (90%) of the instances by standard karyo typing or fluorescent in situ hybridization (FISH) techniques. Variable translocations of heavy or light chains (MYC/IGH or MYC/IGL) may be depicted. The tumour variants in the children and young adults may belong to the germinal centre subtype (CD10+ or BCL6+ & MUM1-) as delineated by the genetic expression profiling (GEP). The network of the genetic mutations elucidated in the lymphoma may comprise of the ID3, TCF3 and CCND3 pathways (50- 90%). The MYC oncogene may implicate the ID3(80%)and TCF3(13%) which may involve the BCR and P13K complexes, affecting the evolution or perpetuation of the tumour cells [2, 6]. The molecule Cyclin D3 (36%) may be incriminated in the conversion of the cell cycle phase from growth to DNA synthesis (G1/S) and cellular proliferation. The cytogenetic evaluation may be beneficial in demarcating betwixt the classic Burkitt’s lymphoma and c MYC associated B lymphocyte predominant non hodgkin’s lymphoma, particularly the diffuse large B cell lymphoma (DLBCL) [2, 6]. The majority of the instances of Burkitt’s lymphoma may be distinguished by a specific karyotype such as a t(8:14)(q24:q32) translocation along with the gain of chromosomes 1q. The adjunctive c MYC induced B lymphocyte non hodgkin’s lymphoma may depict a complicated karyotype with multitudinous chromosomal gains and/or losses. As yet not a usual procedure, however, the microarray based gene expression profiling (GEP) may categorize a Burkitt’s lymphoma with the molecular methodology [2, 5].
Adjunctive genetic adaptations may be truncating mutations in ARID1 A, amplification of MCL1, point mutations of LRP6, truncating amendments of LRP1B, PTPRD, PTEN, NOTCH & ATM along with amplifications of RAF1, MDM4, MDM2, KRAS, IKBKE and CDK6, deletion of CDKN2A, over expression of MIR 17HG, activating mutations of TCF3, CCND 3 and inactivating mutations of ID3.Non reactive instances of Epstein barr virus (EBV) may display several genetic corrections, in contrast to the reactive (EBV+) individuals. Amplification 13q 31:3q32.1, gain of 7q:32:q36 and 5q23.3 copy neutral loss of heterozygosity (CN-LOH) may be detected in young patients. 17p13 and 18q21.3 copy neutral loss of heterozygosity (CN- LOH) may be displayed in adults. ID3 double hit mutations as well as 18q21 copy neutral loss of heterozygosity (CN-LOH) may be associated with a worse prognosis [2, 3].
The aberrant Bukitt’s lymphoma may delineate a predominant and obscuring granulomatous response. The anomalous morphology may be constantly reactive for the Epstein barr virus (EBV+) genome with categorical t(8:14) cytogenetic translocations. The granulomas may probably be a T helper cell response to nuclear viral (EBNA 1) antigens. The nodal and extra-nodal gastro- intestinal tumours may retrogress spontaneously. The interpretation of the tumour may be challenging on account of the granulomas. A particularly favourable outcome may be anticipated with the low grade, limited stage disease. Immature granulomatous configurations may signify an appropriate immune response with spontaneous retrogression of the tumour [2, 6].
Burkitt’s lymphomas as a constituent of AIDS defining illness may be reactive for the virus (EBV+) in one fourth or more (25-40%) instances. The pleomorphism and plasmacytoid delineation may be enhanced with the characteristic immune-phenotype. The disorder needs a distinction from the plasmablastic lymphoma. The tumour may display a MYC translocation, the absence of B cell markers( CD19, CD20, CD34 and IgM) and a post germinal centre phenotype (activated B cells CD10-, MUM1+ , BCL6+/- )(3,5) . Burkitt’s lymphoma may exceptionally account for <5% of the post transplant lympho-proliferative disorder (PTLD). The Epstein barr virus (EBV) may co-exist in up to one third to three fourths(30-70%) instances, delineated by molecular techniques. The variant of Burkitt’s lymphoma may be non-reactive for MYC oncogene. The 11q aberration may be detected in less than half (42%) of the post transplant instances [2].
A contemporary variant with a lack of t(8;14) or diverse translocations along with Burkitt’s lymphoma with duplication 11q23:q13 has been described. A de- regulation of mi R34b may be detected in the MYC negative Burkitt’s lymphoma. Interstitial gains incorporating 11q23.2-q23.3 and telomeric deficiency of 11q24.1 qter may be diagnosed in the molecular Burkitt’s lymphoma besides the adjunctive MYC negative high grade B cell lymphomas and lymphomas simulating Burkitt’s lymphomas. An augmentation of 11q23.3 with over expression of PAFAH1B2 and a deficiency of ETS1 may be encountered. ID3 and TCF 3 mutations may be lacking. The prevalence and the clinical significance of the particular variant is indeterminate which may be frequent in immune deficient patients with a nodal emergence. The histology is identical to Burkitt’s lymphoma, however the monotonous round cells may be variable and pleomorphic. The immune phenotype may resemble a classic Burkitt’s lymphoma with a depreciating MYC expression. The karyotype may be intricate (CNA type). The MYC, ID3, TCF 3 genotypic mutations may be lacking, in contrast to the classic Burkitt’s lymphoma. A Burkitt’s lymphoma which is MYC negative , a high grade B cell lymphoma – not otherwise specified(HG-BCL NOS) with absent double hit mutation , a lymphoma with a classical Burkitt’s phenotype, a post transplant Burkitt’s lymphoma or a paediatric kind of follicular lymphoma, may define the situations where an 11q aberration may be suspected or detected [2, 3].
MYC rearrangements may be elucidated in the Burkitt’s lymphoma (90%), peripheral blood lymphocytes (50%), multiple myeloma (15-50%), diffuse large cell B cell lymphoma (DLBCL 5-10%), high grade B cell lymphoma not otherwise specified (HG BCL NOS 30%- 50%) and with double hit lymphomas (DH 100%) [2, 3].
Sporadic MYC rearrangements may be depicted in the high grade B cell lymphoma (HG-BCL) or the Tdt+ precursor double hit B cell lymphoma (DL BCL) or in follicular lymphomas, t cell follicular lymphomas, chronic lymphocytic leukaemia or in chronic myeloid leukaemia (tFL, tCLL, tCML) [7, 8, 9] Figures 1-11.
Contrasting Diagnoses
Burkitt’s lymphoma may require a distinction from a) The diffuse large cell B cell lymphoma (DLBCL) of the germinal centre subtype (GCB). The tumour cells may be enlarged with a diverse morphology and an immune phenotype of BCL2-, MYC rearrangements (10%) single MYC rearrangements (4%) with non Ig G translocation partner [2].
b) The high grade B cell lymphoma (HG BCL) with double hit or triple hit (MYC/ BCL2/ BCL6) mutations, not otherwise specified (NOS). Diffuse large B cell lymphomas (DLBCL) with MYC + 5-15%, a BCL2 /BCL 6 translocation, the designated double hit and triple hit lymphomas respectively [2, 3].
c) The blastoid variant , previously known as the Burkitt’s
like lymphoma (BLL) with a poor prognosis or the unclassifiable B cell lymphoma (u) with intermediary attributes of diffuse large B cell lymphoma (DLBCL) and Burkitt’s lymphoma [2, 5]. The characteristic therapeutic regimen for the Burkitt’s lymphoma or the diffuse large B cell lymphoma (DLBCL) may not be efficacious. The histological sub-category may include multiple aggressive B cell lymphomas which may not categorically indicate the morphology, immune- histochemical (IHC) or cytogenetic profile and the clinical attributes of a classic Burkitt’s lymphoma.
d) Burkitt like lymphoma with 11q aberration.
e) Precursor lymphoblastic leukaemia/ lymphomas (B-
LBL) with miniature blasts (CD 34+ and Tdt+).
f) Mantle cell lymphoma, particularly the blastoid variant (CD5+, Cyclin D1+, SOX 11+) [2, 6]. Myeloid sarcoma g) Small round cell tumours.
h) Lymphomas with morphology l and immune- phenotype intermediate to Burkitt’s lymphoma and diffuse large B cell lymphoma (DLBCL), ascribed to as “gray –zone lymphomas” [3].
Lymphomas Simulating Burkitt’s Lymphoma
a) Paediatric lymphomas may be diagnosed by morphology akin to Burkitt’s lymphoma and immune phenotype of CD20+, CD10+, BCL6+, BCL2-/w, Tdt -, MYC re-arrangements >80% Ki67 > 95%. It may require a differentiation from the classic Burkitt’s lymphoma [1].
b) Adult Burkitt’s lymphoma may be diagnosed by morphology and immune phenotype such as CD20+, CD10+, BCL6+, BCL2-/w, Tdt -, MYC re-arrangements >80% Ki67 > 95% an absence of double hit mutation,11q abnormality or a discontinuity in the MYC. It may require a demarcation from a Burkitt;s like lymphoma with 11q abnormality [2, 3].
c) Diffuse large B cell lymphoma (DLBCL) with a classical morphology re-categorized as Burkitt’s Lymphoma with genetic expression profiling (GEP) may depict the following molecular modifications: Ki- 67 converging to a 100%, immune phenotype of germinal centre B cells (GCB), BCL-2 non reactive, p53+ reactive , p21- non reactive. MYC rearrangements may be elucidated, BCL2/BCL6 rearrangements may be lacking. The cellular morphology may be of DLBCL. Therapeutic responsiveness to cyclophosphamide, vincrinstine, doxorubicin & methotrexate /ifosfamide with etoposide and cytarabine (CODOX –M/ IVAC) may be delineated [6].
d) Adult Burkitt’s lymphoma with atypical morphology and phenotypic aberrations such as CD10-, MUM1/IRF4++, BCL 2++, MYC < 80%, Ki67< 95% with a presence or an absence of a double hit mutation. It may necessitate a distinction from a high grade B cell lymphoma with a double hit mutation or a high grade B cell lymphoma not otherwise specified (HG–BCL NOS) [2, 3].
e) Irrespective of the age group : A morphology of the Burkitt’s lymphoma and immune phenotype of CD20+, CD10+, BCL6+, BCL2-/w, Tdt -, MYC re-arrangements <80% Ki67 > 95%, an absence of double hit mutation,11q abnormality or a discontinuity in the MYC. It may require a demarcation from a Burkitt;s like lymphoma with 11q abnormality [7, 8].
The paediatric Burkiit’s lymphoma with the implicated adolescent and young adults (AYAs) may be appraised by the Murphy and Hustu’s classification. The common extra- nodal involvement and the inadequate resection of the intra-abdominal or intra-thoracic lumps may be estimated by the system. Adult patients have not been corroborated by the applied classification. The Ann Arbor’s classification may be employed for the prognostic evaluation of the tumour [1, 2, 3] Table 1.
f) Adult lymphoma with a blastoid morphology and an immune phenotype of CD20+, frequently CD10+, BCL6+, BCL2+, consistently Tdt- and CyclinD1- with a presence or absence of double hit mutations. It may necessitate a distinction from a high grade B cell lymphoma with a double hit mutation or a high grade B cell lymphoma not otherwise specified( HG –BCL NOS) [1, 2].
g) Adult diffuse large B cell lymphomas (DLBCL) with classic morphology, immune phenotype with the absence or presence of double hit mutations. It requires a distinction from diffuse large B cell lymphomas- not otherwise specified (DLBCL–NOS) or a high grade B cell lymphoma with a double hit mutation [2, 3].
Analytical Profile
A speedy staging is necessitated in order to institute the therapy within forty eight hours of diagnosis. An individual history, a general physical examination particularly for B symptoms (fever, night sweats, weight loss), the performance status (PS) and signs and symptoms of neurological involvement may be required. Haematological and biochemical parameters such as a complete blood count(CBC) , hepatic and renal functions tests, serum electrolytes, serum uric acid , serum lactic dehydrogenase(LDH) and serology for viral infections including human immune deficiency (HIV), Epstein barr virus( EBV) and hepatitis B and C may be a pre-requisite [1, 3] . Contrast enhanced computerized tomography (CT) of the chest, abdomen and pelvis may be necessary. The positron emission tomography (PET) scan may not be usually employed, even though the tumour cells are 18 fluoro-deoxy glucose (FDG) avid. A bone marrow aspiration with a bone marrow biopsy and a lumbar puncture with flow cytometric analysis of the cerebro spinal fluid (CSF) may be required. Cardiac investigations (trans-thoracic echocardiography), a pregnancy test as appropriate and an assessment with counselling of the fertility concerns may be mandated [1, 2].
Tumour Execution
| Murphy’s System | Ann Arbor System | |||||
|---|---|---|---|---|---|---|
| Stage I | Single nodal or extra-nodal site excluding mediastinum or abdomen | Single nodal or extra-nodal site. | ||||
| Stage II | Two or more nodal areas on one side of the diaphragm. | Two or more nodal areas on one side of the diaphragm. | ||||
| Localized involvement of an extra-lymphatic site and of one or more extra-nodal sites on same side of the diaphragm (IIE). | ||||||
| Stage II R | Complete resection of the intra-abdominal disease | |||||
| Stage III | Two or more nodal areas on opposite side of the diaphragm. | Two or more nodal areas on opposite side of the diaphragm which may include/involve the spleen IIIs | ||||
| Primary intra-thoracic tumour or | or a localized involvement of an extra-nodal site (IIIE) | |||||
| Para-spinal or epidural tumours | ||||||
| Extensive intra-abdominal disease | ||||||
| Stage IIIA | Localized intra-abdominal disease not amenable to resection | |||||
| Stage IIIB | Widespread multi organ intra-abdominal disease | |||||
| Stage IV | Central nervous system or bone marrow involvement | Diffuse or disseminated involvement of one or more extra-lymphatic sites | ||||
| Two single extra-nodal tumours on the opposite side of the diaphragm | ||||||
| Also A and B depending upon symptoms. | ||||||
| Favourable | Stage I or IIR | Stage IA and IIA | ||||
| Stage I,II and III |
Table 1: Contemporary Categorization by Murphy and Ann Arbor [1].
The prognostic sub-stratum of various therapeutic groups may segregate the patients by virtue of the age, performance status (PS), serum lactic dehydrogenase (LDH)levels, the bone marrow findings and the symptoms of central nervous system (CNS) involvement [1, 6]. The LYSA GRALL intergroup may identify three prognostic sub-groups: i) Group A: constitutes of patients with a comprehensive resection and individuals with stage I or abdominal stage II disease( Murphy’s classification).ii) Group C includes individuals with infiltration of the central nervous system (CNS)and the bone marrow. ii) Group B incorporates patients which do not classify for groups A or C. The DANA FARBER intergroup may be collated into two prognostic sub-groups [1]. i) The low- risk sub-group indicates individuals with a normal serum lactic dehydrogenase (LDH), an appropriate performance status (PS-ECOG 0 or1) and a tumour mass <10centimetres. ii) The high risk sub-group includes patients who do not qualify as low risk. The MD Anderson cancer centre along with the Cancer and Leukaemia The determinants may coordinate the magnitude, duration and the class of the therapeutic regimen (drug combinations, the numerical score of treatment cycles of the systemic and intra-thecal chemotherapy and the requirement of radiotherapy) [9, 10]. The chemo- sensitivity of the tumour is considered the optimal prognostic indicator [1, 2]. The delineation of minimal disseminated disease (MDD) at diagnosis may indicate a subclinical or sub microscopic quantity of lymphoma/ leukaemia cells. Minimal residual disease (MRD) may be elucidated following appropriate therapy in patients with complete clinical response [6]. An enhanced lactic dehyrogenase (LDH), primary mediastinal involvement, combined bone marrow /central nervous involvement, co-existent viral (HIV+) infection, deletion of 13q, gain of 7q with a complicated cytogenetic disease may depict a poor prognosis. Double hit mutations with genetic aberrations of ID3 and/or CCND3 with 18q21 copy neutral loss of heterozygosity (CN-LOH) may enunciate a sub-minimal therapeutic response and an unfavourable prognosis [2, 6] Table 2.
| mBL | Non- mBL | |||||
|---|---|---|---|---|---|---|
| Age>60 years | 9% | 69% | ||||
| Cell of Origin | ||||||
| Germinal centre B cell | 91% | 35% | ||||
| Activated B cell | 39% | |||||
| Unclassified | 9% | 26% | ||||
| BCL2 expression | 21% | 84% | ||||
| Proliferation Index (>95%) | 66% | 12% | ||||
| MYC translocation | ||||||
| IG MYC | 88% | 4% | ||||
| Non IG MYC | 2% | 3% | ||||
| Translocation absent | 9% | 93% | ||||
| IGH-BCL2 | 2% | 11% | ||||
| BCL-6 translocation | 24% | |||||
| Genomic complexity | ||||||
| Low | 79% | 21% | ||||
| High | 21% | 79% | ||||
| Genetic groups | ||||||
| MYC simple | 76% | |||||
| MYC complex | 13% | 7% | ||||
| MYC negative | 11% | 93% |
Table 2: The molecular categorization of Burkitt’s Lymphoma [6].
Tumour Lysis Syndrome
A colossal cytolysis accompanied by effusion of
phosphates and chelated calcium ions leading to the
precipitation within the renal proximal and distal
convoluted tubules may ensue in the tumour lysis
syndrome. An elevation in the serum uric acid, potassium,
phosphates and hypocalcaemia is characteristic. Acute
renal impairment, disruption of the cardiac rhythm,
dysfunction or arrhythmia, seizures on account of the
toxicity of the central nervous system and symptomatic
hypocalcaemia may arise. The laboratory investigations of
$$ \text {hypocalcemia may arise. The laboratory investigations of the disorder may incorporate} \geq \text {two metabolic} $$
abnormalities with serum uric acid >8 mg/dl, potassium >
6.0 mmol /l, phosphates >4.5mg/dl, total serum calcium
<7.0gm/dl with ionized <1.12mg/dl and an elevated
serum creatinine. Pre-existing renal impairment, an
elevated pre-treatment uric acid and lactic
dehydrogenase (LDH), male gender with splenomegaly
Therapeutic Re-Assessment
A computerized tomography (CT) scan may be required halfway and at the termination of the treatment. The residual lumps may be evaluated by a positron emission tomography (PET) scan. Following the first year of therapeutic assistance, the computerized tomography
Dictums of Therapy
Treatment should be instituted within forty eight hours following detection. The tumour lysis syndrome (TLS) should be prohibited with appropriate hydration, diuresis and administration of rasburicase. Cycles of chemotherapy may be briefly interrupted. The patients lacking human immune deficiency virus (HIV-) infection may be managed by dose adjusted etoposide, prednisone, vincristine, cyclophophamide and doxorubicin with rituximab ( DA-EPOCH R) [11, 12]. Individuals with the viral infection (HIV+) may adopt the short cycle etoposide, prednisone, vincristine, cyclophophamide and doxorubicin with riuiximab (SC-EPOCH RR) [11].
Therapeutic Guidelines
Intensive short cycle chemotherapy (ISCC) inclusive of cyclophosphamide, cell cycle phase specific agents that move across the blood brain barrier along with cytarabine and methotrexate may be beneficial. The extension of the tumour into the central nervous system may be prohibited with the application of intrathecal methotrexate, cytarabine, and prednisone [13]. Low intensity chemotherapy (LIC) with dose adjusted (DA- EPOCH R) etoposide, vincristine, prednisone, cyclophosphamide, doxorubicin and rituximab may be applicable along with Short Cycle etoposide, vincristine, prednisone, cyclophosphamide, doxorubicin and rituximab (SC EPOCH R)(!4). The adjunct treatment cycle may commence within the shortest possible period. The employment of colony stimulating factors (G-CSF) may be mandatory. An autologous stem cell transplant may be efficacious in partial response (PR) [14].
Intensive short cycle chemotherapy (ISCC) maybe beneficial for the LYSA GRAAL intergroup, the German Multicentre Study Group for the Adult ALL (GMALL) or the Dana Farber intergroup with an appropriate benefit / toxicity ratio. A therapeutic combination with highly active anti-retroviral therapy (HAART) may enhance the therapeutic response and survival percentages [1]. The recently detected Burkitt’s lymphoma of the young individuals (AYAs) may be managed with the intensive short cycle chemotherapy(ISCC) regimen The healthy, elderly patients up to age 75 years of age may be similarly treated by modifying the doses of cytarabin and methotrexate [1, 6]. Patients beyond 75 years with or those < 75years with co-morbidities and a lack of central nervous system (CNS) involvement may benefit from the dose adjusted regimen with rituximab (DA-EPOCH R). Human immune deficiency virus (HIV+) infected patients lacking co-morbidities may be managed by intensive short cycle chemotherapy (ISCC). Individuals with a poor performance status (PS) and negligible central nervous system involvement may benefit from a short cycle regimen (SC-EPOCH R) [14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25]. The antiCD20 monoclonal antibodies of ofatumumab and obinutuzumab, the anti CD19 molecules blinatumomab and the anti CD 22 antibodies inotuzumab, ozogamicin are the contemporary addition to the armamentarium of the therapeutic agents necessitating extended evaluation [3].
Response to Therapy
The therapeutic response may be assessed by the complete remission (CR), partial response, stable disease, progressive disease when established lesions may amplify to more than half (> 50 %) in magnitude or recent, current lesions may merge. Residual nodules which are nonreactive by the positron emission tomography (PET) scan may be cogitated as a complete remission (CR). A false positive emission scan may signify a benign inflammatory lesion, a xanthoma or a pseudo-tumour, uptake of brown fat , rejuvenating thymic hyperplasia, infection or the aftermath of the administered colony stimulating factors(G-CSF) [1, 2].
References
-
Bonnet C, Janssens A, Wu KL, Schroyens W, Van Hende V, et al. (2015) BHS guidelines for the treatment of Burkitt’s lymphoma. Belgian J Haematol 6(2): 61-69.
-
Chris Bacon Recent advances in the genetics and biology of lymphomas” Northern Institute of Cancer Research, Newcastle University 2017: Newcastle Upon Tyne Hospitals NHS Foundation Trust.
-
Twenty ninth European Congress of Pathology: pathology for patient care: Burkitt’s lymphoma and variant” Sep 2017 Amsterdam.
-
Rosai and Ackerman’s ‘Surgical Pathology’ 10th (Edn.), pp: 1842.
-
Dozzo M, Carobolante F, Donisi PM, Scattolin A, Maino E, et al. (2017) Burkitt’s lymphoma in adolescents and young adults: management challenges. Adolescent Health Med Ther 8: 11-29.
-
Steven H Swerdlow, Elias Campo, Stefano A Pileri, Nancy Lee Harris, Harald Stein, et al. (2016) The 2016 revision of world health organization classification of lymphoid neoplasm BLOOD 127(20): 2375-2390.
-
Michael J Kluk, Bjoern C, Papiya S, Alyssa R, Paola Dal Cin, et al. (2012) Immune histochemical detection of the MYC driven diffuse large B cell lymphoma. Plos one e33813.
-
Li S, Lin P, Fayad LE, Lennon PA, Miranda RN, et al. (2012) B cell lymphoma with MYC 18q24 rearrangements and IGH@ BCL2/t(8:14)(q32:q21): an aggressive disease with heterogeneous histology/ germinal centre B cell immune phenotype and poor outcome” Mod Pathol 25(1): 145-156.
-
Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, et al. (2012) Burkitt’s Lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490(7418): 116-120.
-
Intermesoli T, Rambaldi A, Rossi G, Delaini F, Romani C, et al. (2013) High cure rates in Burkitt’s lymphoma and leukaemia a northern Italy leukaemia group study of the German shot intensive rituximab- chemotherapy program. Haematologica 98(11): 1718-1725.
-
Ahmed SO, et al. (2013) The role of haematopoietic stem cell transplantation in adult Burkitt’s lymphoma. Bone marrow transplants 48: 617-629.
-
Dunleavy K, Stefania P, Margaret Shovlin RN, Seth M Steinberg, Diane C, et al. (2013) Low intensity therapy in adults with Burkitt’s lymphoma. N Engl J Med 369: 1915-25.
-
Petrich AM, Sparano JA, Parekh S (2012) Paradigm and Controversies in the treatment of HIV related Burkitt’s Lymphoma. Adv Hematol 2012: 403648.
-
Nowakowski GS, LaPlant B, Macon WR, Reeder CB, Foran JM, et al. (2015) Lanalidomide combined with R-CHOP overcomes negative prognostic impact of a non-germinal centre B cell phenotype in newly
-
Image 11Courtesy: Haematological Malignancy Diagnostic Services.com diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol 33(3): 251-257.
-
Nowakowski GS, Blum KA, Kahl BS, Friedberg JW, Baizer L, et al. (2016) Beyond R –CHOP: A blueprint for diffuse large B cell lymphoma research. J Natl Case Instt 108 (12): 257.
-
Image 1 Courtesy: Pathology Outlines.
-
Image 2 Courtesy: Webpathology.com
-
Image 3 Courtesy: Ganfyd.com
-
Image 4 Courtesy: Research gate
-
Image 5 Courtesy: uaz.edu. mx
-
Image 6 Courtesy: Pathology outlines.
-
Image7 Courtesy: Pinterest.com
-
Image 8 & 9 Courtesy: Wikimedia common
-
Image 10Courtesy: openi.nlm.nih.com
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies