Hematopoietic Stem Cell Transplantation: An Approach in the Treatment of Sickle Cell Disease
Hematopoietic Stem Cell Transplantation (HSCT) is a treatment indicated for various types of hematological neoplasms, such as leukemia, lymphoma, multiple myeloma, and other autoimmune hematological diseases and immunodeficiencies. Sickle cell disease (SCD) is an inherited blood disorder that shortens the survival of red blood cells, causing anemia. This research sought to address advances and new perspectives in HSCT in the treatment of sickle cell disease. Currently, HSCT is the only curative option for patients with sickle cell disease and aims to restore normal hematopoiesis and thus prevent damage from successive episodes of sickling. Allogeneic HSC transplantation has become an increasingly acceptable treatment option for sickle cell disease. Currently, even with advances in supportive therapy to prevent complications of SCD, access to care for this disease is unequal. However, worldwide, 120 million people are affected by the disease, and of these, 66% live in Africa. Therefore, in Africa and Asia, the number of patients tends to grow. Therefore, it is important to apply and expand neonatal screening programs, and the need for investment in health policies, especially in these countries where the disease is more severe.
Introduction
Hematopoietic Stem Cell Transplantation (HSCT), sometimes called Bone Marrow Transplantation (BMT), is a treatment indicated for several types of hematological neoplasms, such as leukemia, lymphoma, multiple myeloma, and other autoimmune hematological diseases and immunodeficiencies [1].
During the last two decades, the use of this therapy has expanded throughout the world and its technology has evolved. Nowadays, it is used for new indications, such as autoimmune and hereditary metabolic disorders [2, 3].
Pediatric HSCT presents unique challenges, such as the need to adjust doses of chemotherapy and radiotherapy according to the child’s age and size. Furthermore, emotional and psychological support for both the child and the family is essential throughout the process, as the transplant and the recovery period can be intense and challenging [4].
Sickle cell disease (SCD) is an inherited blood disorder that shortens the survival of red blood cells, causing anemia. According to the World Health Organization (WHO), approximately 5% of the world’s population carries trait genes for hemoglobin disorders, mainly sickle cell disease and thalassemia. Annually, more than 300,000 babies are born with serious hemoglobin disorders. Worldwide, 120 million people are affected by the disease, and of these, 66% live in Africa [5].
Considered as the only treatment capable of curing sickle cell anemia, hematopoietic stem cell transplantation (HSCT) is effective in curative therapy for the disease. In this sense, this research sought to address advances and new perspectives in hematopoietic stem cell transplantation (HSCT) in the treatment of sickle cell disease.
Pathophysiology and clinical manifestations of sickle cell disease
Sickle cell disease (SCD) is an umbrella terminology that represents a group of autosomal recessive diseases. There are variable genotypes of SCD, but the most common is sickle cell anemia (HbS), which results from the inheritance of two copies of the HbS mutation (homozygous) and accounts for 65% to 70% of all cases of SCD [6].
Sickle cell disease (SCD) is hereditary resulting from an autosomal recessive genetic mutation of red blood cells, which results from the inheritance of two sickle cell genes, with one gene from each parent. When both parents carry a gene for hemoglobin S (HbS), known as sickle cell trait, there is a 25% chance of their children inheriting the disease, a 25% chance of their children being unaffected, and a 50% chance of carrying the genetic mutation as an asymptomatic carrier [7].
The disease is characterized by a mutation in the DNA of chromosome 11, caused by the specific replacement of the nitrogenous base thymine (T) with adenine (A), which changes from GAG to GTG [8]. The exchange of nitrogenous bases in DNA (Figure 1), instead of encoding the production (transcription) of the amino acid glutamic acid, will, from then on, determine the production of the amino acid valine, which will enter position 6 in the sequence of amino acids that make up the beta chain of hemoglobin, modifying its molecular structure [9].
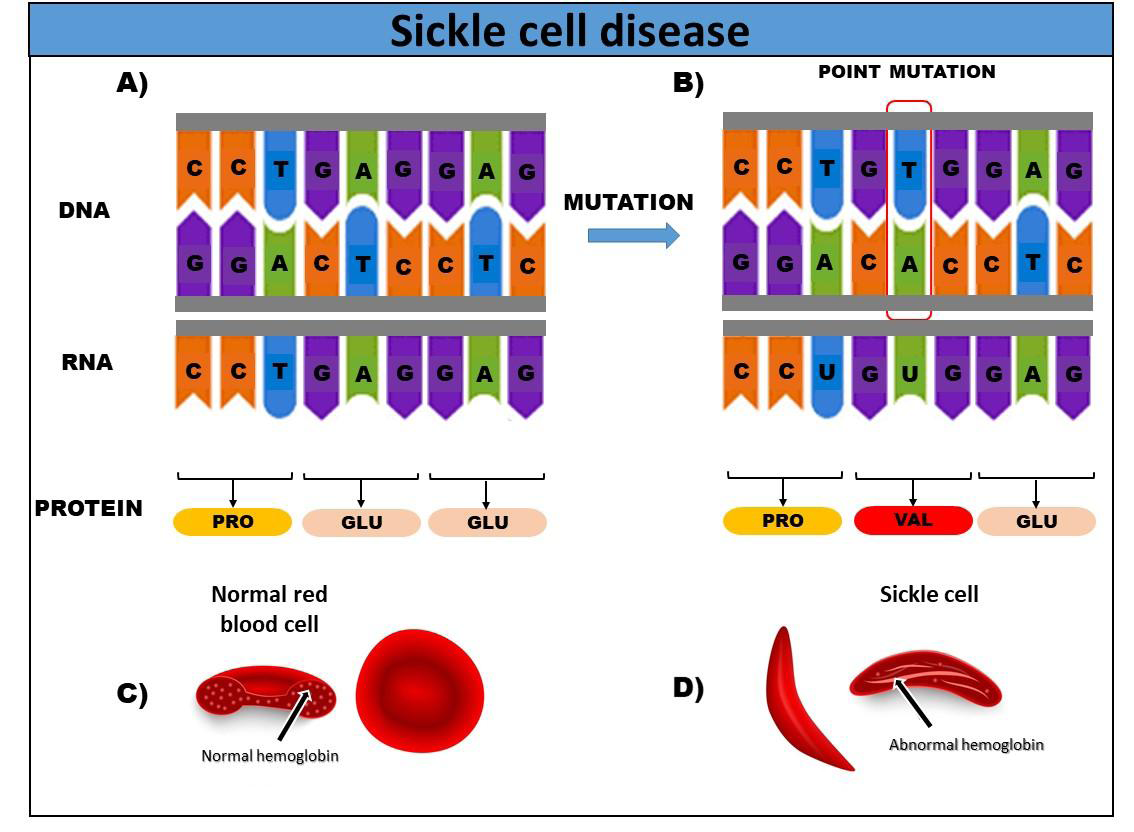
The pathophysiology of sickle cell disease is related to the ability of hemoglobin S to crystallize under conditions of low oxygen concentration. This crystallization leads to deformation of the red blood cells, which take on a sickle or crescent shape instead of their normal disc shape. Sickle red blood cells are less flexible and have a shorter lifespan, which results in chronic anemia [4].
In sickle cell disease (SCD), sickle hemoglobin (HbS) tends to polymerize under conditions of low oxygen saturation. Red blood cells containing HbS polymers tend to convert the normal biconcave disc shape of red blood cells into a rigid and irregular sickle shape that can block blood vessels in the microcirculation, thus impairing the supply of oxygen to tissues [10].
Hematopoietic Stem Cell Transplantation in Sickle Cell Disease
HSCT is the replacement of diseased or suppressed bone marrow with another healthy bone marrow from a histocompatible donor [11]. Currently, hematopoietic stem cell transplantation (HSCT) is the only curative option for patients with sickle cell disease and aims to restore normal hematopoiesis and, thus, prevent damage from successive episodes of sickling [12].
Hematopoietic stem cell transplantation (HSCT) is equivalent to a treatment modality seen as a highly invasive and highly complex procedure, which involves the use of medications chemotherapy, radiotherapy sessions, blood transfusions and other methods that can cause numerous risks to the patient’s life [13]. The treatment consists of providing the patient with progenitor cells that can be taken from the patient himself (autologous transplant), from a donor (allogeneic transplant) or even from umbilical cord cells [1].
The new transplanted cells are intended to reconstitute the patient’s hematopoietic and immune systems. Thus, the production of blood cells and cytotoxic destruction of remaining diseased cells of the recipient are expected to occur. With the success of the transplant, hematopoiesis becomes subordinate to the new graft and no longer to the diseased bone marrow [14].
Allogeneic hematopoietic stem cell transplantation has become an increasingly acceptable treatment option for sickle cell disease. An analysis of 1,000 human leukocyte antigen (HLA)-matched identical sibling donor transplants performed between 1986 and 2013, at 106 centers in 23 countries worldwide. Patients were mainly children (n = 846; <16 years), with a median age of 9 years. The excellent outcome over 3 decades confirms the role of HLA-identical sibling transplantation for children with SCD [15].
Currently, even with advances in supportive therapy to prevent complications of sickle cell disease (SCD), access to care for this disease is unequal. With recent advances in medical treatment, the natural history of sickle cell disease (SCD) continues to evolve as morbidity and mortality decline [16].
Since 1990, 4 large randomized trials to prevent central nervous system (CNS) injury have been completed, presenting evidence-based guidelines for the primary and secondary prevention of stroke in children with SCD [17, 18]. However, in sub-Saharan African countries and India, where more than 90% of children are born with SCD [19] evidence- based primary and secondary stroke prevention strategies are not presented. Thus, ∼50% of children with SCD will have a cerebral infarction before they are 18 years of age.
Allogeneic HSCT, graft-versus-host disease (GVHD) is the leading cause of morbidity and mortality in pediatric patients. This disease develops due to a “protection” that the recipient’s immune system carries out against the donor’s cells and, depending on the intensity, can result in rejection of the transplanted graft, which can be acute (most common), occurring in up to 6 weeks after transplantation, or chronic, occurring after 6 weeks of transplantation [20, 21, 22].
In the study by Hsieh, et al. [23], the findings indicate that patients who underwent hematopoietic stem cell transplantation (HSCT) mostly presented a reversal of the sickle cell disease phenotype, in addition, those grafted continued to be disease-free and without the manifestation of sickle cell disease. graft versus host, that is, tolerance to the donor cells occurred, which brings greater survival and quality of life to patients.
In a more recent study, de Costa, et al. [24], identified better disease-free survival results after HSCT in children with sickle cell disease, especially with hematopoietic progenitor cells. The findings agree with previous studies [25]. which state that patients undergoing transplantation can have average survival and disease-free survival rates above 90% and 73%, respectively.
Allogeneic HSCT procedure from siblings remains the type of treatment considered the gold standard for the disease, as it has high success rates. However, it is still a therapeutic modality that has the disadvantage of low availability of compatible donors and the risk of developing graft-versus-host disease [26].
Marques, et al. [27] state that, even though hematopoietic stem cell transplantation is the hope of many patients, representing, in most cases, the only form of treatment for cases of SCD, it still has serious implications such as the occurrence of SCD. Acute graft versus host, causing impacts on the quality of life of these patients, in their psychological, social, biological aspects and cognitive impairment in children and adolescents, according to Castro, et al. [28].
The quality of life of transplanted children with sickle cell disease can also be harmed by the appearance of early and late complications, such as the evolution of insulin resistance, systemic arterial hypertension and/or dyslipidemia and, consequently, the development of metabolic syndrome after HSCT [29].
Hematopoietic stem cell transplantation (HSCT) can cure children and adults with sickle cell disease. Outcomes have historically been poor for the vast majority of patients who do not have a matching sibling donor. However, the development of haploidentical HCT (haplo-HCT) with high doses of post-transplant cyclophosphamide (PTCy) has allowed long-term curative potential with favorable transplant-related outcomes, although this has not prevented the potential for graft rejection of the human leukocyte antigen incompatibility and repeated red blood cell transfusions [26].
Conclusions
The present study sought to investigate the current perspectives on hematopoietic stem cell transplantation in the treatment of sickle cell disease. In this research it can be observed that sickle cell disease is a highly prevalent pathology in children and adults, however, in recent years, there have been advances in diagnosis and treatment, however, it is not a reality in poor countries such as Africa and Asia, where the number of patients tends to grow. In this sense, it is important to apply and expand neonatal screening programs, and the need for investment in health policies, especially in those countries where the disease is more severe.
References
-
Magedanz L, Leal JVO, Santos BL, Brito ES, Saavedra PAE, et al. (2022) Hematopoietic Stem Cell Transplantation: Inequities in Distribution in Brazilian Territory, 2001 to 2020. Public Health Science 27 (08), 3239-3247.
-
Daikeler T, Hügle T, Farge D, Andolina M, Gualandi F, et al. (2009) Allogeneic Hematopoietic SCT for Patients with Autoimmune Diseases. Bone Marrow Transplantation 44(1): 27-33.
-
Hirano M, Marti R, Casali C, Tadesse S, Uldrick T, et al. (2006) Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology 67(8): 1458-1460.
-
Brunetta DM, Cle DV, Haes TM, Roriz-Filho JS, Moriguti JC (2010) Management of Acute Complications Of Sickle Cell Disease. Medicine (Ribeirão Preto) 43(3): 231-237.
-
WHO (2024) African health ministers launch drive to curb sickle cell disease toll.
-
Egesa WI, Nakalema G, Waibi WM, Turyasiima M, Amuje E, et al. (2022) Sickle Cell Disease in Children and Adolescents: A Review of the Historical, Clinical, and Public Health Perspective of Sub-Saharan Africa and Beyond. International Journal of Pediatrics.
-
National Heart, Lung, and Blood Institute (2023) Sickle Cell Disease Causes and Risk Factors.
-
Ware RE, Montalembert M, Tshilolo L, Abboud MR (2017) Sickle Cell Disease. The Lancet 390(10091): 311-323.
-
Sundd P, Gladwin MT, Novelli EM (2019) Pathophysiology of Sickle Cell Disease. Annual Review of Pathology: Mechanisms of Disease 14: 263-292.
-
Wang Q, Zennadi R (2021) The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants (Basel, Switzerland) 10(10): 1608.
-
Silva AF, Kosmaliski ULC, Antunes BS, Motta MGC (2022) Allogeneic Hematopoietic Stem Cell Transplantation in Children and Adolescents: Ethical Problems Faced by the Multidisciplinary Team. Rev. Gaucha Enferm 43: e20210315.
-
Barban JB, Simões BP, Moraes BDGC, Anunciação CR, Rocha CS, et al. (2020) Brazilian Consensus on Nutrition in Hematopoietic Stem Cell Transplantation: Adults. Einstein (São Paulo) 18: AE4530.
-
Thomson B, Gorospe G, Cooke L, Giesie P, Johnson S (2015) Transitions of Care: A Hematopoietic Stem Cell Transplantation Nursing Education Project across the Trajectory. Clinical Journal of Oncology Nursing 19(4): E74-E79.
-
Zago MA, Falcão RP, Pasquini R (2004) Hematology: fundamentals and practice. Ed the Atheneu, São Paulo, pp: 1081.
-
Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, et al. (2017) Sickle Cell Disease: An International Survey of Results of HLA-Identical Sibling Hematopoietic Stem Cell Transplantation. Blood 129(11): 1548-1556.
-
DeBaun MR, Kirkham FJ (2016) Central Nervous System Complications and Management in Sickle Cell Disease. Blood 127(7): 829-838.
-
Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, et al. (1998) Prevention of a First Stroke by Transfusions in Children with Sickle Cell Anemia and Abnormal Results on Transcranial Doppler Ultrasonography. N Engl J med 339(1): 5-11.
-
Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, et al. (2016) Hydroxycarbamide Versus Chronic Transfusion for Maintenance of Transcranial Doppler Flow Velocities in Children with Sickle Cell Anaemia-TCD with Transfusions Changing to Hydroxyurea (Twitch): A Multicentre, Open-Label, Phase 3, Non-Inferiority Trial. Lancet (London, England) 387(10019): 661-670.
-
Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN (2013) Global Burden of Sickle Cell Anaemia in Children Under Five, 2010-2050: Modelling Based on Demographics, Excess Mortality, And Interventions. PLOS Medicine 10(7): e1001484.
-
Lee SJ, Onstad L, Chow EJ, Shaw BE, Jim HSL, et al. (2018) Patient-Reported Outcomes and Health Status Associated with Chronic Graft-Versus-Host Disease. Haematologica 103(9): 1535-1541.
-
Punnett A, Sung L, Price V, Das P, Diezi M, et al. (2007) Achievement of Target Cyclosporine Concentrations as a Predictor of Severe Acute Graft Versus Host Disease in Children Undergoing Hematopoietic Stem Cell Transplantation and Receiving Cyclosporine and Methotrexate Prophylaxis. Therapeutic Drug Monitoring 29(6): 750-757.
-
Wilhelm AJ, Graaf P, Veldkamp AI, Janssen JJ, Huijgens PC, et al. (2012) Population Pharmacokinetics of Ciclosporin in Haematopoietic Allogeneic Stem Cell Transplantation with Emphasis on Limited Sampling Strategy. British Journal of Clinical Pharmacology 73(4): 553-563.
-
Hsieh MM, Fitzhugh CD, Weitzel RP, Link ME, Coles WA, et al. (2014) Non-myeloablative HLA-Matched Sibling Allogeneic Hematopoietic Stem Cell Transplantation for Severe Sickle Cell Phenotype. JAMA 312(1), 48-56.
-
Costa TCM, Darrig-Junior LG, Grecco CES, Pieroni F, Stracieri ABPL, et al. (2023) Allogenic Hematopoetic Progenitor Cell Transplantation for the Treatment of Sickle Cell Disease: Results from a Single Brazilian Center. Hematology, Transfusion and Cell Therapy 45(4): S528.
-
Freed J, Talano J, Small T, Ricci A, Cairo MS (2012) Allogeneic Cellular and Autologous Stem Cell Therapy for Sickle Cell Disease: ‘Whom, When and How’. Bone Marrow Transplantation 47(12): 1489-1498.
-
Patel DA, Akinsete AM, Fuente JL, Kassim AA (2020) Haploidentical Bone Marrow Transplant with Posttransplant Cyclophosphamide for Sickle Cell Disease: An update. Hematology/Oncology And Stem Cell Therapy 13(2): 91-97.
-
Marques ACB, Szczepanik AP, Machado CAM, Santos PND, Guimarães PRB, et al. (2018) Hematopoietic Stem Cell Transplantation and Quality of Life During the First Year of Treatment. Rev. Latino-Am. Enfermagem 26: e3065.
-
Castro IPS, Viana MB (2019) Cognitive Profile of Children with Sickle Cell Anemia Compared to Healthy Controls. Journal of Pediatrics 95(4): 451-457.
-
Ozenen GG, Aksoylar, Goksen S, Gozmen D, Darcan S, et al. (2021) Metabolic Syndrome and Risk Factors after Hematopoietic Stem Cell Transplantation in Children and Adolescents. Journal of Pediatric Endocrinology and Metabolism 34(4): 485-493.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies