Management of Delayed Hemolytic Transfusion Reaction in a Patient with Sickle Cell Disease: A Case Report
Background: Sickle cell disease (SCD) is a group of inherited blood disorders characterized by abnormalities in hemoglobin (Hb), leading to distortion in shape of red blood cells (RBCs) and their breakdown, potentially blocking blood flow and resulting in pain and anemia. This can significantly impact the patient’s quality of life. Patients often require multiple blood transfusions to prevent complications associated with SCD by reducing the concentration of damaged hemoglobin. However, one of the significant complications of blood transfusions is Delayed Hemolytic Transfusion Reaction (DHTR), which occurs due to RBC alloimmunization (i.e., formation of antibodies against donor RBC antigens). DHTR can manifest as a sudden decrease in hemoglobin levels, which may progress to rapid multiorgan failure and death. Thus, prompt diagnosis is crucial for timely management. This case report highlights the diagnosis and management of a 56-year-old African American female with SCD who presented with bilateral severe knee pain and low Hb levels. Case Presentation: The patient’s symptoms, past medical history, and investigation findings led to a diagnosis of DHTR. Management was conducted within the framework of Bloodless Medicine, incorporating strategies to minimize blood loss and optimize erythropoiesis. Treatment included the use of erythropoietin (EPO) to manage hemoglobin levels and the administration of opioids and supplemental oxygen for symptom relief. Conclusion: Our case report highlights the challenges and various management strategies employed in treating DHTR in SCD patients. It emphasizes the importance of early recognition of the complication and compares the risks and benefits of additional blood transfusions in such patients. By presenting this case, we aim to contribute to the growing literature on DHTR in SCD patients and highlight the need for further research and optimized management strategies in this population.
Introduction
Sickle cell disorder (SCD) is an autosomal recessive multiorgan disorder caused by a point mutation in the beta- globin chain of hemoglobin that results in substitution of the hydrophobic amino acid valine for glutamic acid. The disease severity varies from sickle cell trait (HbAS), which is a milder form, to sickle cell disease (HbSS) depending on the number of defective alleles inherited [1].
As a global pathology, the disease can be found in sub- Saharan Africa, Spanish-speaking regions on the Western side of the world (South America, the Caribbean, and Central America), the Middle East, South Asia and Mediterranean countries. Approximately 100,000 Americans live with SCD; the incidence is 1 out of 365 and 1 out of every 16,300 in the African American population and Hispanic American births, respectively; whereas 1 in 13 African-American babies are born with Sickle Cell Trait (SCT) [2].
SCD can have a wide range of clinical presentations and complications, both acute and chronic. This is mostly due to vaso-occlusive crisis resulting in severe organ damage notably affecting the brain, lungs, spleen and skeleton, further aggravating the morbidity and mortality caused by the disease [3, 4].
As a multisystem disorder, it is difficult to diagnose on the basis of clinical manifestations. The diagnosis of SCD is based on the detection of Hemoglobin S (HbS) in relation to Hemoglobin A (HbA) and Hemoglobin F (HbF). The commonly used methods are hemoglobin electrophoresis, high-pressure liquid chromatography, isoelectric focusing and molecular approaches such as PCR [5, 6].
The neutrophil-to-lymphocyte ratio (NLR) is a reliable inflammatory biomarker in SCD, with significant prognostic implications across various SCD states [7, 8]. Mean platelet volume (MPV) reflects platelet size diversity and may serve as an early predictor of cerebrovascular events in sickle cell anemia (SCA) [9, 10]. Red cell distribution width (RDW) is a simple and readily available inflammatory biomarker, with a cutoff point of 19.9 at a 95% confidence interval, proving effective for screening SCA phenotypes [11, 12].
As SCD affects multiple organ systems, a multidisciplinary team approach is needed that not only educates patients and family members but also guides them regarding the treatment of a lifelong disease and increases access to psychological and social support.
RBC transfusion constitutes a crucial treatment for both acute and chronic manifestations of SCD, including the prevention of stroke [13]. It can be administered in three ways-
- Simple Transfusion- Simple transfusion involves the transfusion of packed red blood cells without removing any of the patient’s blood volume.
- Chronic Transfusion Therapy- Chronic transfusion therapy involves simple transfusions on schedule of every two to four weeks. This therapy may be used for stroke prevention for patients at high risk for stroke.
- Exchange Transfusion- Exchange transfusion is the removal of a percentage of the patient’s blood and replacement with donor packed red blood cells.
This is a much more time-consuming process and is usually performed in the dialysis area of the hospital using an apheresis machine. This method substantially reduces the concentration of sickle cells without increasing the overall hematocrit or blood viscosity (thickness).
One of the most concerning complications of RBC transfusion in the SCD population is red cell alloimmunization (development of clinically significant antibodies in the recipient against donor RBC antigens); 19–47% of SCD patients are alloimmunized, more than any other patient group [13]. This can lead to DHTR which is a potentially life-threatening condition. We describe a case of DHTR in a patient with Sickle Cell Disease in our case report [14].
Case Presentation
We present a case of sickle cell crisis in a 56-year-old African American female who presented with bilateral severe knee arthralgia (10/10 intensity) refractory to her home medication (hydrocodone-acetaminophen). She had a history of sickle cell disease, delayed hemolytic transfusion reaction due to non-ABO incompatibility, hydroxyurea intolerance, pulmonary hypertension, chronic kidney disease with sickle cell nephropathy, Salmonella osteomyelitis in the left tibia and fibula, transfusion related iron overload, and nontoxic multinodular goitre.
She had previously been admitted to the hospital for an episode of sickle cell crisis complicated by pneumonia, for which she received Ceftriaxone + Azithromycin and was discharged 3 days before the current hospital admission. Her home medications included Apixaban, Ergocalciferol, Losartan, Folic acid, hydrocodone-acetaminophen and oxybutynin.
The patient’s family history was noncontributory.
On physical examination, her vitals were stable except for her oxygen saturation level of 94%. She was alert and oriented to time, place and person. She was in moderate distress due to pain in her knees.
Investigations
Investigations at the time of admission revealed anemia with Hemoglobin level of 5.1 g/dL and an RBC count of 2.31 million/mm3. She also had a moderate number of sickle cells, moderate polychromasia, schistocytosis, anisocytosis, microcytosis, hypochromia, marked poikilocytosis, slight ovalocytosis and dacrocytosis.
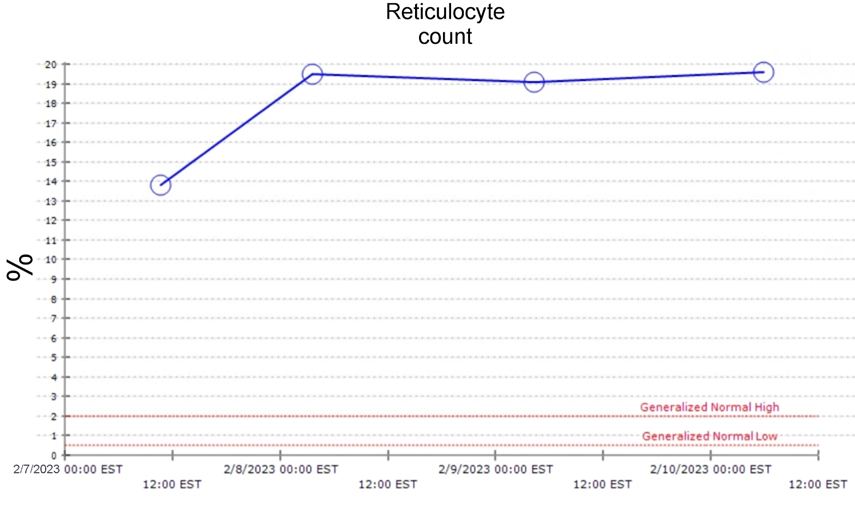
Her reticulocyte count was 13.8% (high) with a high Red Cell Distribution Width (RDW) of 19.3%. Her baseline hemoglobin level was approximately 6.0 g/dL. Some of her pertinent test results are mentioned below in Table 1.
| Laboratory Parameter | Value on day of Admission (February 7, 2023) |
|---|---|
| Complete Blood Count | |
| WBC (White Blood Cells) | 19.9 thousand/mm3 |
| RBC (Red Blood Cells) | 2.31 million/mm3 |
| Hemoglobin | 5.1 g/dL |
| Hematocrit | 16.70% |
| MCV (Mean Corpuscular Volume) | 72.1 fL |
| MCH (Mean Corpuscular Hemoglobin) | 22.1 pg |
| MCHC (Mean Corpuscular Hemoglobin Concentration) | 30.7 g/dL |
| RDW (Red Cell Distribution Width) | 19.30% |
| Platelet | 563 thousand/mm3 |
| Reticulocytes | 13.80% |
| Absolute Reticulocyte Count | 5.5 cells/µL |
| Iron Studies | |
| Iron level | 85 µg/dL |
| % of iron saturation | 32% |
| Total Iron Binding Capacity | 262 µg/dL |
| Ferritin | 3160 ng/mL |
| Transferrin | 183.12 µg/dL |
| Vitamin B12 level | 272 pg/mL |
| Renal and Liver Function Tests | |
| BUN (Blood Urea Nitrogen) | 14 mg/dL |
| Creatinine | 0.91 mg/dL |
| Total Bilirubin | 2.7 mg/dL (high) |
| Direct Bilirubin | 0.6 mg/dL |
| LDH (Lactate Dehydrogenase) | 826 U/L (high) |
| eGFR (estimated Glomerular Filtration Rate) | 74 mL/min/1.73m2 |
| Other Pertinent Test Results | |
| ABO/Rh Interpretation | A positive |
| Antibody Screen Interpretation | Negative |
| Direct Coombs’ Test (IgG C3d) | Negative |
Table 1: Investigation findings of the patient.
Radiological Findings
Her chest X-ray (Figure 1) revealed central vascular prominence with right lower lung and left mid and lower lung opacities suggestive of scarring.
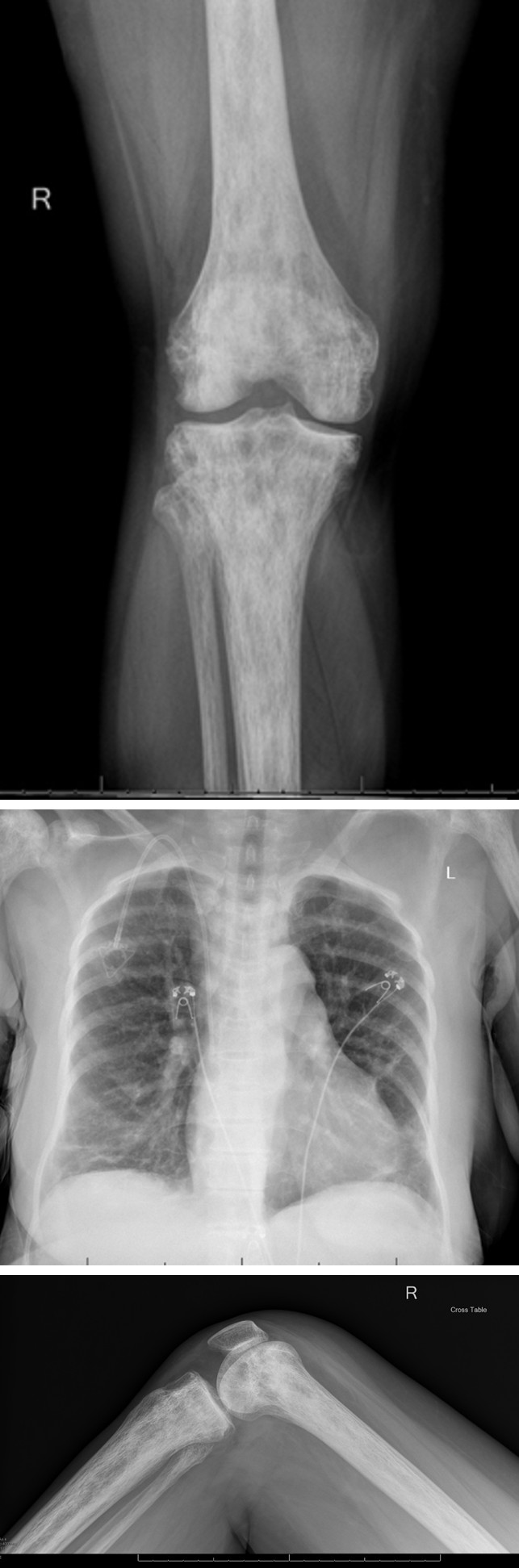
Additionally, her right knee X-ray (Figures 2A and 2B) was performed to evaluate the causes of her knee pain. The X-ray showed no acute fracture or malalignment, maintained joint spaces, and patchy areas of sclerosis involving the visualized femur and proximal tibia/fibula, likely representing bone infarcts given the provided history of sickle cell disease. There was no definitive radiographic evidence of osteomyelitis or any suprapatellar joint effusion.
Figure 2A: Right knee X-ray of the patient (Lateral view).
Figure 2B: Right knee X-ray of the patient (Anteroposterior view).
CT angiography of the chest was also performed to rule out pulmonary embolism. There was no CT evidence of pulmonary embolism, pneumonia, or thoracic aortic dissection. There was interval development of bibasilar opacities within the dependent portions of the lower lungs, likely suggestive of acute chest syndrome, along with chronic occlusion of the brachiocephalic vein.
Diagnosis
This patient was diagnosed with a hemolytic crisis due to sickle cell disease, as evidenced by her low hemoglobin levels; along with a vaso-occlusive pain crisis due to which she was experiencing pain in her right knee. Other causes of low hemoglobin levels in sickle cell patients including Splenic Sequestration Crisis (usually seen in young children with SCD, while in older patients autoinfarction of spleen occurs), and Aplastic Crisis (due to Parvovirus B19, which causes a decrease in reticulocyte count), were ruled out [15].
A potential cause of the sudden decrease in hemoglobin could be the Packed Red Blood Cell (PRBC) transfusion during her previous hospital admission, further supporting the diagnosis of delayed hemolytic transfusion reaction (DHTR). Treatment The patient was placed on the Bloodless Medicine pathway and monitored for judicious transfusion. Her EPO level was 1806.
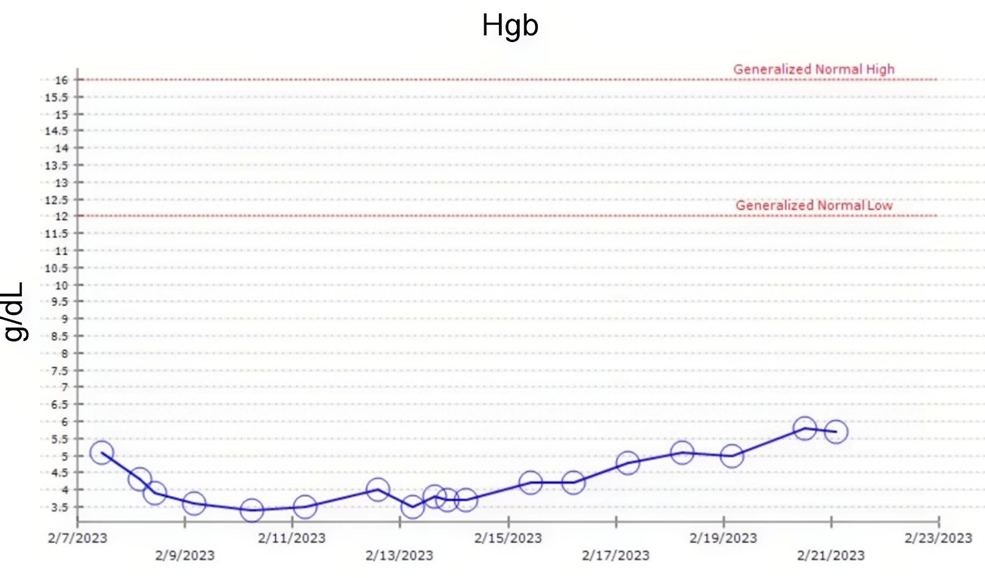
She was started on EPO at a dose of 40,000 U daily. However, throughout the course of her hospital admission, her Hemoglobin levels ranged between 3.5 g/dL and 5.1 g/ dL, and finally increased to 5.8 g/dL at the time of discharge (Day 15).
Research conducted on erythropoiesis stimulating agents (ESAs) with the goal of enhancing haemoglobin levels in SCD patients has yielded conflicting results. Some studies that focused solely on ESAs have shown improvements in anaemia or a reduced need for blood cell transfusions, while others have not. When ESAs were given to SCD patients who were already receiving HC treatment some studies demonstrated an increase in haemoglobin concentrations, while others showed mixed or no clear positive effects. There is limited data on the outcomes of starting ESA treatment simultaneously with HC. In addition, two of these studies have raised concerns about ESAs triggering vasoocclusive crises (VOCs) [16]. EPO use, especially for Hb levels greater than 11 to 12 g/dL, was associated with an increased risk of vascular access clotting and cerebrovascular events [17]. She was given 40 mg Enoxaparin daily to counter this dreaded calamity throughout the course of her hospital admission.
Daily B12 was continued for low normal B12 level. The patient’s blood draws were also minimized, to minimize her blood loss and to prevent a further decrease in her Hemoglobin. This was in accordance with the approach of Bloodless Medicine, which is an increasingly recognized and accepted concept in Haematology.
Her Haemoglobin levels throughout the course of her hospital admission are shown in Figure 3 (all the dates mentioned are presented in Month/Day/Year format).
Her reticulocyte count remained high throughout the course of her hospital admission, which is represented graphically in Figure 4 (all the dates mentioned are presented in Month/Day/Year format.
Other medications, such as opioids, antibiotics, losartan and supplemental oxygen, were continued for symptomatic management.
This management regimen was continued with regular monitoring until her Haemoglobin level improved to over 5 g/dL, and she was discharged on Day 21 on her home EPO.
Discussion
DHTR is defined as a significant decrease in haemoglobin within 21 days posttransfusion associated with 1 or more of the following: new red cell alloantibody, hemoglobinuria, accelerated HbS% increase with a concomitant decrease in HbA, relative reticulocytopenia or reticulocytosis from baseline, significant LDH increase from baseline, and exclusion of alternative causes [18]. However, DHTR should be quickly suspected in any SCD patient with sudden worsening of clinical and laboratory signs of haemolysis within two weeks of a blood transfusion. Maintaining a high index of suspicion for DHTR is crucial because mistakenly diagnosing another complication (e.g., acute chest syndrome, sickle cell crisis, aplastic crisis etc.) may lead to further transfusions, which can exacerbate haemolysis [19].
The incidence of DHTR in SCD patients ranges from 0.001% to 13.52%, with mortality rates ranging from 6% to 11%, suggesting significant implications for SCD mortality rates [20]. The pathophysiology of DHTR in SCD patients is unclear. DHTR is believed to be caused by anti- RBC antibodies attacking donor RBCs, but these antibodies are often not detected in these patients. DHTR can occur even after transfusions that are matched for the ABO, Rhesus (Rh), and Kell blood antigen systems and after a negative serological cross match test. Other hypotheses include accelerated eryptosis (erythrocyte apoptosis) and inflammatory enzymes causing RBC destruction.
The risk factors for DHTR in patients without detectable antibodies are unknown, making it challenging to identify those at risk for a first or recurrent episode [19]. While most SCD patients receive a blood transfusion at some point in their lifetime, not every patient develops alloimmunization or DHTR, which suggests that there are case-specific factors at play. Possible risk factors include high transfusion burden (although it does not strongly correlate with the development of alloimmunization), genetic factors related to B lymphocyte signal modulation, differences in T cell population, recipient inflammatory state at the time of transfusion (SCD patients transfused during a proinflammatory complication such as Acute Coronary Syndrome appear to be at increased risk for subsequent RBC alloantibody development), and environmental factors [21, 22]. One study reported that differences in RBC antigen expression between the SCD patients receiving PRBC transfusions (mainly African Americans, in whom the disease is more common) and the donors (mainly Caucasians) may contribute to the greater likelihood of alloimmunization in SCD patients [21].
Diagnosis of DHTR can be difficult based on haemolysis markers, particularly in patients with SCD who experience chronic haemolysis [20]. Early diagnosis of DHTRs is possible based on pain and hemoglobinuria, which are noted in 95% and 89% of patients, respectively. Patients should be educated about these symptoms, and emergency room clinicians should maintain the possibility of DHTRs as a differential for recently transfused SCD patients with concerning findings. Repeated haemoglobin concentration measurements can confirm whether transfused RBCs are being destroyed. Early diagnosis is essential to prevent paradoxically harmful blood transfusions and guide appropriate treatment [23].
The unclear pathogenesis and difficult diagnosis of DHTR in SCD patients account for its difficult management, which varies according to the clinical severity, and ranges from supportive care and optimization of erythropoiesis to immunosuppression [24]. Recommendations for DHTR treatment include stopping further transfusions; optimizing erythropoiesis; addressing common hematopoietic deficiencies by administering iron, folic acid, vitamin B12, and EPO; and use of immunosuppressive therapy (if required), including IVIG (Intravenous Immunoglobulin), high dose steroids, rituximab, and eculizumab [19, 20].
Avoiding further transfusion is usually recommended, but transfusion may be done after extended RBC antigen matching (which includes C/c, E/e, K, Jka/Jkb, Fya/Fyb, and S/s) if patients are experiencing life-threatening anaemia with on-going haemolysis [18].
Hydroxyurea (which increases HbF production) may be considered in SCD patients who are un-transfusable to maximize haemoglobin levels and reduce the need for transfusions in the long term [20].
The principles of Bloodless Medicine can be helpful in managing such patients. Bloodless Medicine employs strategies such as minimization of blood loss (e.g., by judicious use of phlebotomy and use of appropriate pharmacologic agents), optimization of blood production, use of autologous blood transfusions and advanced bloodless surgical techniques, to treat patients without the need for allogeneic blood transfusions [25]. This approach is similar to the approach employed while treating Jehovah’s witnesses [26]. The concept of Bloodless Medicine can provide a new direction for managing DHTR in SCD patients with severe anaemia.
The American Society of Haematology (ASH) guidelines suggest immunosuppressive therapy (IVIG, steroids, and/ or rituximab) over no immunosuppressive therapy in patients with SCD (all genotypes) with an acute need for transfusion and at high risk for acute hemolytic transfusion reaction, those with a history of multiple or life-threatening delayed hemolytic transfusion reactions, or those having a delayed hemolytic transfusion reaction with on-going hyperhemolysis [18]. Immunosuppressive therapy should be initiated promptly in patients with life-threatening haemolysis. High-dose steroids and IVIg are considered first- line treatments, followed by eculizumab for patients who continue to experience clinical deterioration despite first- line agents. Rituximab is primarily indicated for potential prevention of additional alloantibody formation in patients who may require further transfusion. Depending on the length of steroid therapy, weaning should be considered to avoid precipitation of a vaso-occlusive episode. Supportive care, including erythropoietin with or without IV iron, should be initiated in all patients. Discussion between a haematologist and a transfusion medicine specialist is advised to determine the optimal approach for each patient [18].
The use of methylprednisolone or prednisone at 1 to 4 mg/kg per day and IVIG at 0.4 to 1 g/kg per day for three to five days (up to a total dose of 2 g/kg) demonstrated effective results in a study. Similarly, eculizumab used in adult patients (>40 kg) at a weekly dose of 900 to 1200 mg, for hyperhemolysis, and rituximab at a dose of 375 mg/m2 with repeat dose after 2 weeks, have also shown promising outcomes. IVIG competitively blocks mononuclear phagocytes in patients with anti-RBC antibodies; however, it may have different modes of action in patients without detectable antibodies. It also alters the cellular immune response by affecting lymphocyte and monocyte apoptosis and cytokine modulation, and induces the production of Interleukin-1 receptor antagonist (which inhibits the phagocytosis of donor RBCs by recipient macrophages and reduces inflammation) [24]. Steroids and IVIG have been found to work synergistically to suppress macrophage activity [20]. Eculizumab, a C5 inhibitor, was found to be successful in treating a SCD patient with severe DHTR, according to a recent case report by Nickel et. al. However, standardised clinical trials need to be conducted to assess its efficacy before it can be used as a standard treatment [22]. Swee, et al. suggest that excessive complement activation may account for severe DHTR presentations with accompanying hyperhemolysis (where recipient cells undergo hemolysis and haemoglobin levels fall to levels lower than pretransfusion). These cases may benefit from eculizumab, which might prevent irreversible multi-organ failure and lead to rapid clinical improvement [20]. The patient discussed in our case report was not a candidate for immunosuppression as she did not present with life threatening haemolysis.
Additional clinical trials are required to explore the most effective dosages, potential side effects, expenses, and rates at which patient outcomes improve when immunosuppressants and supportive drugs are used. ASH also recommends conduction of high-quality studies to understand the mechanisms and consequences of alloantibody-mediated clearance of transfused red cells, and determine the efficacy of immunomodulatory agents in DHTRs [18]. Discovering better ways to prevent and manage RBC alloimmunization is the need of the hour because recent evidence suggests that long term RBC transfusions could benefit a large number of SCD patients, but this currently comes at the cost of increasing the risk of alloimmunization [22]. Establishing an international DHTR registry with extensive clinical and biological parameters can also lead to new research directions for identifying risk factors, triggers, and mechanisms of this complex post-transfusion reaction [23].
Conclusion
Our case report highlights the challenges and strategies involved in the management of DHTR in a patient with SCD. It emphasizes the importance of early recognition and diagnosis of DHTR and the need for individualized treatment plans to address the unique challenges that patients with SCD have to face. Bloodless Medicine principles aimed at minimizing blood loss and optimizing erythropoiesis offer a promising direction for managing severe anemia in SCD patients while avoiding allogeneic blood transfusions. Immunosuppressive therapies are effective in the management of such patients, but the use of ESAs in SCD patients remains a topic of debate, with conflicting results and multiple risks.
Our case report contributes to the growing body of knowledge on DHTR management in SCD patients and highlights the need for further research and clinical trials to enhance our understanding and improve treatment strategies.
Data Availability
All data underlying the results are available as part of the article and no additional source data are required.
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this article.
Funding Statement
No funding was received to assist with the preparation of this case report.
Acknowledgements
Jessica Houpe- Resident, Dermatology
References
-
Quinn CT (2016) Minireview: Clinical severity in sickle cell disease: the challenges of definition and prognostication. Exp Biol Med (Maywood) 241(7): 679- 688.
-
(2023) Centers for Disease Control and Prevention. Data and Statistics on Sickle Cell Disease.
-
Inusa BPD, Hsu LL, Kohli N, Patel A, Evbota K, et al. (2019) Sickle Cell Disease Genetics, Pathophysiology, Clinical Presentation and Treatment. International Journal of Neonatal Screening 5(2): 20.
-
Mangla A, Ehsan M, Agarwal N, Smita M (2023) Sickle Cell Anemia. StatPearls.
-
Arishi WA, Alhadrami HA, Zourob M (2021) Techniques for the Detection of Sickle Cell Disease A Review. Micromachines (Basel) 12(5): 519.
-
Acharya B, Mishra DP, Barik B, Ranjan Kumar M, Ashish Kumar S (2023) Recent progress in the treatment of sickle cell disease: an up-to-date review. Beni-Suef Univ J Basic Appl Sci 12(1).
-
Emokpae M, Aruomaren A, Osime E (2016) Relationship between neutrophil-to-lymphocyte ratio and inflammatory markers in sickle cell anaemia patients with proteinuria. Med Sci (Basel) 4(3): 11.
-
Uslu AU, Kucuk A, Sahin A, Ugan Y, Yilmaz R, et al. (2015) Two new inflammatory markers associated with disease activity score-28 in patients with rheumatoid arthritis: neutrophil-lymphocyte ratio and platelet lymphocyte ratio. Int J Rheum Dis 18(7): 731-735.
-
Hameed M, Abozied M (2013) Mean platelet volume in impaired fasting Glucose subjects and diabetic patients as a risk factor for thrombotic complications. J Am Sci 9(9): 12-17.
-
Celik T, Unal S, Ekinci O, Ozer C, Ilhan G, et al. (2015) Mean platelet volume can predict cerebrovascular events in patients with sickle cell anemia. Pak J Med Sci 31(1): 203-208.
-
Hammed MR, Tohamy MA, Boshra SZ, Taha SM, Saleh MFM (2023) Red cell distribution width is an inflammatory predictor marker of contrast induced nephropathy in patients undergoing percutaneous coronary intervention. Egypt J Immunol 30(3): 1-12.
-
Mutua B, Sowayi G, Okoth P (2022) Prognostic potential of RDW in discriminating hemoglobinopathies among patients reporting to Aga Khan Hospital, Kisumu. Egypt J Med Hum Genet 23(1).
-
Annika M, Meghan D (2015) Considerations of red blood cell molecular testing in transfusion medicine. Expert Rev Mol Diagn 15(11): 1455-1464.
-
Scheunemann LP, Ataga KI (2010) Delayed hemolytic transfusion reaction in sickle cell disease. Am J Med Sci 339(3): 266-269.
-
Borhade MB, Kondamudi NP (2023) Sickle Cell Crisis. In StatPearls [Internet]. Treasure Island (FL).
-
Han J, Zhou J, Kondragunta V, Zhang X, Molokie RE, et al. (2018) Erythropoiesis-stimulating agents in sickle cell anaemia. Br J Haematol 182(4): 602-605.
-
Parfrey PS (2006) Target hemoglobin level for EPO therapy in CKD. Am J Kidney Dis 47(1): 171-173.
-
Chou ST, Alsawas M, Fasano RM, Field JJ, Hendrickson JE, et al. (2020) American Society of Hematology 2020 guidelines for sickle cell disease transfusion support. Blood Adv 4(2): 327-355.
-
Montalembert M, Dumont MD, Heilbronner C, Brousse V, Charrara O, et al. (2011) Delayed hemolytic transfusion reaction in children with sickle cell disease. Haematologica 96(6): 801-807.
-
Alwaheed AJ, Alqatari SG, AlSulaiman AS, Sulaiman RS (2022) Delayed hemolytic transfusion reaction in sickle cell disease A case series. Am J Case Rep 5(23): e934681.
-
Higgins JM, Sloan SR (2008) Stochastic modeling of human RBC alloimmunization evidence for a distinct population of immunologic responders. Blood 112(6): 2546–2553.
-
Nickel RS, Hendrickson JE, Fasano RM, Meyer EK, Winkler AM, et al. (2016) Impact of red blood cell alloimmunization on sickle cell disease mortality a case series. Transfusion 56(1): 107-114.
-
Habibi A, Mekontso DA, Guillaud C, Michel M, Razazi K, et al. (2016) Delayed hemolytic transfusion reaction in adult sickle cell disease presentations outcomes and treatments of 99 referral center episodes DHTR in SCD Patients. Am J Hematol 91(10): 989-994.
-
Gardner K, Hoppe C, Mijovic A, Thein SL (2015) How we treat delayed haemolytic transfusion reactions in patients with sickle cell disease. Br J Haematol 170(6): 745-756.
-
Shander A, Goodnough LT (2006) Objectives and limitations of bloodless medical care. Curr Opin Hematol 13(6): 462-470.
-
(2023) Joint United Kingdom (UK) Blood Transfusion and Tissue Transplantation Services Professional Advisory Committee. Jehovah’s Witnesses and blood transfusion.
- How to Identify and Overcome Barriers in Developing Blood Systems?
- Why Was Transfusion Medicine Not Recognized as a Clinical Discipline?
- Outcomes of Lenalidomide Relapsed/Refractory Patients
- Is Transfusion Always Necessary?
- The Logistics of Production and Use of Blood and Blood Components
- The Challenge for Component Therapies