The Morphological Effect of Cisplatin Drug on Cancer Cells. Implications for Breast Cancer Treatment
Triple-negative breast cancer (TNBC) is a proliferating form of breast cancer where there are no oestrogen, progesterone or HER2 receptors, and it comprises about 10–20% of all breast cancer. TNBC also has higher rates of recurrence, increased metastatic potential and is harder to treat because there are no targeted therapies available. Chemotherapy especially agents such as cisplatin remains a foundation of TNBC treatment, but chemoresistance is a clinical issue. This paper reports the cytotoxic and anti-migratory activity of cisplatin on MCF-7 and MDA-MB-231 breast cancer cell lines and the emergence of cisplatin resistance. We also report that longer-term cisplatin treatment drastically reduces cell viability, and IC50 values decrease from approximately 1M after 24 hours to 0.08 M after 48 hours. The cytotoxicity of cisplatin also slows cell migration (the wound closure of MDA-MB-231 cells decreases by 40% in response to cisplatin). But the rise of cisplatin-resistant phenotypes and accompanying morphological changes indicating EMT points towards targeted strategies to overcome resistance. These findings point to cisplatin’s dual cytotoxic and anti-metastatic action and point to resistance mechanisms being addressed to enhance the outcome of TNBC patients
Mohamed M A E L Salem* and Dauda AM
Abbreviations
BC: Breast Cancer; EMT: Extracellular Matrix; MDR: Multidrug Resistance; TNBC: Tripple Negative Breast Cancer.
Introduction
Breast cancer (BC) continues to be a serious public health problem globally in both High-Income countries as well as Middle and Low-income countries, in part because of its high incidence and mortality rates (Breast Cancer. org). Breast cancer is still the world’s leading cause of cancer morbidity and mortality, accounting for 2.3 million new cases and 685,000 deaths in 2020 [1, 2]. Out of all its subtypes, triple-negative breast cancer (TNBC) is the most arduous to treat because it is aggressive, more recurrent, and offers a very narrow therapeutic range because it lacks oestrogen receptor (ER), progesterone receptor (PR), and HER2 expression Burstein et al.
Although TNBC is only detected in about 10-20 per cent of all BC cases, its greater aggressiveness and lack of therapeutic targets, due to the lack of oestrogen, progesterone, and HER2 receptors, have stirred up a sense of urgency. TNBC is known to be more likely to recur, metastasise, and be chemo resistant. Unfortunately, there are currently no FDA-approved targeted cancer therapies for TNBC, and resistance to chemotherapy tends to develop rapidly. This understanding of the molecular reasons for the emergence of chemoresistance would be useful for the development of novel therapies that can address this problem [3].
Cisplatin is a platinum chemotherapeutic drug, which is the main treatment modality for TNBC because it causes DNA to crosslink, causing death. But it can be easily undermined by the inexorable progression of chemoresistance through, for example, increased drug efflux, repair of DNA damage, and EMT [4]. Chemotherapy remains one of the mainstays of TNBC treatment but is often compromised by the development of multidrug-resistance (MDR) [5], a complex multifactorial process that is mediated mainly by drug target alterations with more efficient drug efflux, DNA damage repair, and Epithelial-mesenchymal transition (EMT) [4] and Housman et al. RAN, a small GTPase involved in the regulation of nucleocytoplasmic transport, has recently emerged as a major promoter of tumorigenicity and metastatic dissemination, as well as a key contributor to chemoresistance in different cancers including colon cancer [6], and breast cancer [7]. A recent review also identified RAN GTPase, another nucleocytoplasmic transporter, in tumour growth and chemoresistance in many types of cancers, including breast cancer. RAN levels are increased in aggressive tumours and worse prognoses, so it is an excellent candidate for treatment [8]. RAN is overexpressed in many humans, and represents a poor prognostic factor of the disease, with higher levels found in aggressive and metastatic [9, 10]. One aim of this study is to investigate the response of BC cell lines to cisplatin, a commonly used chemotherapeutic drug, to understand drug sensitivity and propensity to the development of resistance. This study will support the development of novel therapeutic strategies that lead to improved response in patients with BC by rendering cancer cells more sensitive to chemotherapy.
Our aim was to compare cisplatin effects in MCF-7 and MDA-MB-231 breast cancer cell lines, in terms of cytotoxicity, migration and resistance, in order to inform the development of new strategies to optimize therapy for TNBC.
Materials and Methods
Cell Culture
The cell lines chosen were two epithelial cell lines, namely breast cancer cell lines MCF-7 [11]. and MDA-MB- 231(The MDA-MB-231 cell line, isolated at M D Anderson from a pleural effusion of a patient with invasive ductal carcinoma is commonly used to model late-stage breast cancer. This cell line is ER, PR, and E-cadherin negative and expresses mutated p53. In microarray profiling, the MDA-MB-231 cell genome clusters with the basal subtype of breast cancer. Since the cells also lack the growth factor receptor HER2, they represent a good model of triple- negative breast cancer. These cell lines were used because of their importance in cancer research and their divergence in levels of aggressiveness and drug sensitivities. Cells were grown in T75 canted-neck tissue culture flasks in a complete Dulbecco’s Modified Eagle’s Medium (DMEM) with CO2, containing 10 percent fatal bovine serum (FBS), and penicillin streptomycin (1 percent, pen/strep) at 37°C in 5 percent CO2 for humidity. MCF-7 and MDA-MB-231 cell lines were chosen as models for hormone receptor-positive breast cancer and triple-negative breast cancer, respectively. These cancer cell models are known to be divergent in terms of hormone receptor expression, aggressiveness, and drug sensitivities.
In Vitro Cell Viability Assay
The viability of cells was assessed by CCK-8 assay. CCK-8 detected cellular viable metabolic activity through cleavage of water-soluble tetrazolium salt WST-8 by cellular esterases and the subsequent production of formazan dye absorbing light at 450 nm. In the GBM xenograft the cells seeded into 96-well plates at a density of 1 × 104 cells per well in 100 µl of medium, followed by incubation for 24 h at 37°C. The treated cells with increasing doses of cisplatin (15nM– 4000nM) for an additional 24 or 48 hours were added after 24 h of incubation. Untreated cells were used as a negative control. Then the 10 µl CCK-8 solutions was added to each well and incubated for 3 h at 37°C. Followed by incubation at 37°C for 3 hours. Absorbance was measured at a wavelength of 450 nm using a microplate reader (Fluostar Omega, BMG Lab Tech GMBH and Germany). The % of cell viability was determined using the following equation:
(A-B) Proliferation % = 100 (C-B) ×
Where A is the absorbance of treated cells with specific concertation of each compound, B is the absorbance of the blank and C is the absorbance of untreated cells. Dose- response curves were plotted and the IC50 values (the concentration of the drug causing 50 % inhibition in the cell growth) were calculated. All experiments were designed as triplicates.
Resistance Cells Development
First, we generated drug-resistant cell lines using our two baselines MCF-7 and MDA-MB-231 using the method described by Shatha and Malek. Briefly, cells were incubated with low levels of cisplatin to start with, and these concentrations of cisplatin were increased every week with a replacement of fresh medium containing the new concentration until cell lines resistant to high levels of cisplatin grew. We showed that these cells can grow in the presence of cisplatin concentrations that reduce the growth of normal cells.
In Vitro Cell Migration Assay
The migration ability of cell lines was estimated using the scratch wound-healing assay. MCF-7 and MDA-MB-231 cells were seeded into 6-well plates and grown until reaching confluence. A uniform scratch was made through the entirety of the cell sheet using a pipette tip, and the cells were then treated with cisplatin at the corresponding IC50 concentrations. Images were taken at time points 0 hours, 3 hours, 6 hours, and 16 hours after treatment followed by measurement of the wound area by calculating the gap width at each time point. The migration percentage was calculated as a change in gap width relative to the initial gap width at T0, where cells were considered untreated and set as 100 percent.
Results
In Vitro Cell Viability Assay
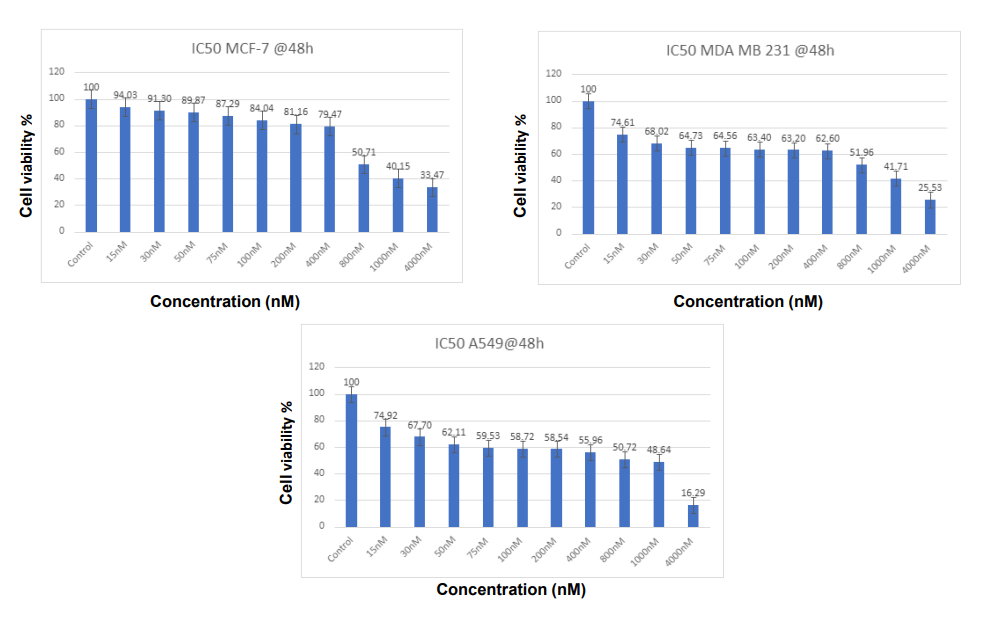
As the cell viability assay shows, the sensitivity of cisplatin for the MCF-7 and MDA-MB-231 breast cancer cell lines is also differential in time. The IC50 values of MCF-7 and MDA-MB-231 cells at 24 hours were around 1 M, which indicated mild drug sensitivity. Yet after 48 hours, IC50 fell drastically to about 0.08 M for both cell lines. And this cumulative sensitivity is consistent with the hypothesis that longer-term cisplatin treatment increases cytotoxic activity, possibly because the damage to DNA can overwhelm cellular repair systems. These results show that cisplatin is highly cytotoxic with longer-term treatment, with cell viability systematically declining as the concentration of cisplatin increases (Figures 1 & 2). As is the case with the aggressive triple-negative breast cancer cells MDA-MB-231, the cells that became more resistant and aggressive, lowered their viability over 24 hours somewhat more slowly than MCF-7 cells [2, 12, 13].
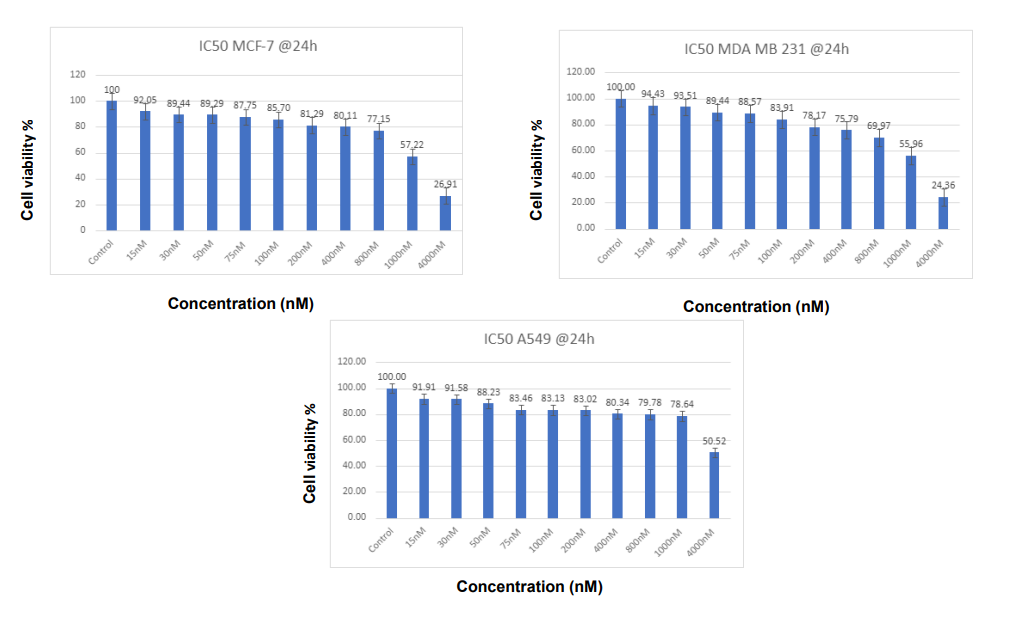
Figure 1: Percentage of cell viability on MCF-7 and MDA-MB-231 cell lines treated with IP-6 Cisplatin - 24 hours. After 24 hours of treatment with Cisplatin, the cell viability of MCF-7 and MDA-MB-231 cells was calculated. It’s clear from Figure 1 that the Viability of breast MCF-7 and MDA-MB-231 cells decreased in contact with increasing dosages of Cisplatin, which is evidence of its effectiveness in fighting cancer. IP-6 has anti-cancer properties that are particularly effective against MCF-7 cancer cells. The increasing activity of IP-6 demonstrates the stronger anti-cancer efficacy at larger dosages.
for 48 hours. This experiment was repeated three times, and for each concentration mean of the percentages was calculated.
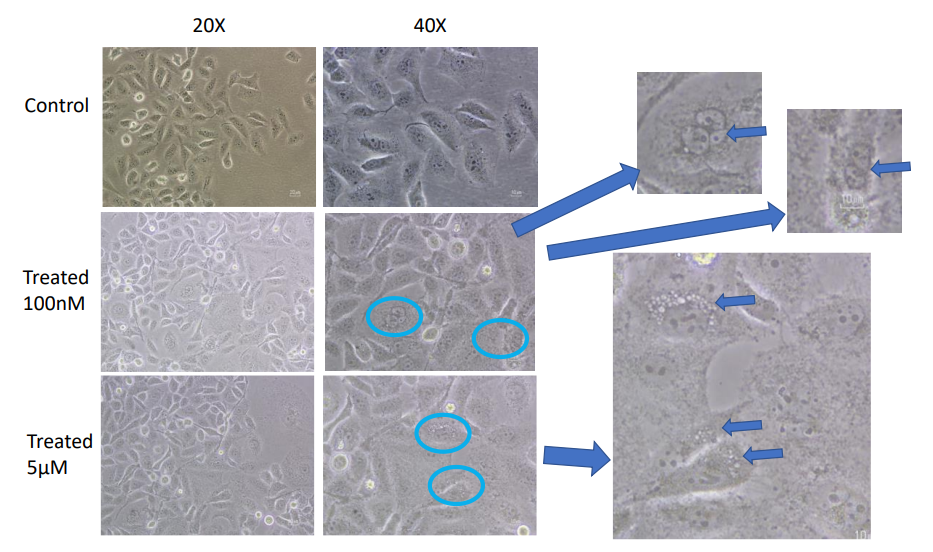
Resistance Cells Development
The growth of cisplatin resistance in both cell lines is shown in Figures 3 & 4. During extended drug use, MCF-7 cells were resistant at a dose of 5 M cisplatin, and MDA- MB-231 cells were resistant at a dose far lower than this, 100 nM. Morphologically, resistant cells were larger and longer, showing epithelial-mesenchymal transition (EMT). This change chimes with the widely known idea that EMT helps cancer cells become more mobile, invasive and resistant [14, 15].
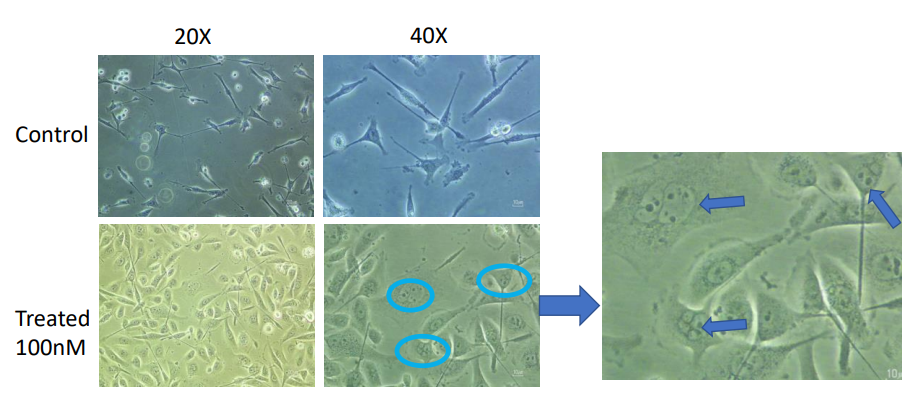
In Vitro Cell Migration Assay
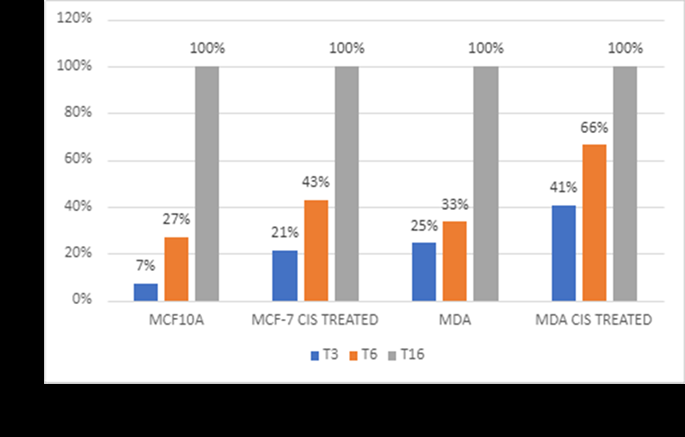
Migration assays also back cisplatin’s inhibitory effect on breast cancer. In untreated controls, MCF-7 and MDA- MB-231 cells were very migrating, and scratch marks were closed quickly by MDA-MB-231 cells due to their aggressive nature. On the other hand, cells treated with cisplatin were much less migrating (Figures 5 & 6). MCF-7 cells treated with 5µM cisplatin and MDA-MB-231 cells treated with 100 µM cisplatin decreased wound closure rates dramatically at 3 (3, 6, and 16 hours). According to these findings, cisplatin slows cell migration, which can dampen metastatic potential [16], consistent with the aggressive, high-motility phenotype of triple-negative breast cancer.
These results support cisplatin’s potential role not only as a cytotoxic agent but also as a suppressor of metastatic potential through the inhibition of cell migration. The observed migration reduction suggests that cisplatin treatment might effectively prevent the spread of breast cancer, particularly in highly metastatic TNBC.

Discussion
The findings of this study provide valuable insights into the differential responses of breast cancer, MCF-7, and MDA-MB-231 cell lines to cisplatin treatment and their resistance. Cisplatin has long been established as a potent chemotherapeutic agent, primarily through its DNA cross- linking activity that induces apoptosis in cancer cells. However, as observed here, responses to cisplatin vary significantly among cancer types, suggesting that intrinsic cellular properties and molecular pathways contribute to differing drug sensitivities.
Enhanced Sensitivity with Prolonged Cisplatin Exposure
This study reveals that there are two ways in which cisplatin works against breast cancer: it causes cytotoxicity, and it blocks metastatic potential. These results are especially notable in the case of triple-negative breast cancer (TNBC), one of the most challenging subtypes of breast cancer to treat because it’s so aggressive and hasn’t received targeted therapies [12]. This striking decline in IC50 after 48 hours of treatment is proof of how critical exposure duration is to get the best benefit from cisplatin. The higher sensitivity found in both cell lines suggests that chronic use of the drug overpowers defence systems, such as DNA repair [2, 13]. This is particularly true of the MDA-MB-231 cells, which are innately more recalcitrant and threatening. Results suggest that cisplatin doses should be adjusted more frequently to optimise treatment results in TNBC.
Mechanisms and Morphological Indicators of Drug Resistance
The gradual development of resistance in MCF-7, and MDA-MB-231 cells to cisplatin supports the hypothesis that cancer cells can undergo adaptive changes under chronic chemotherapeutic pressure. Morphological shifts observed in resistant cells, such as increased size and elongated shapes, suggest that epithelial-mesenchymal transition (EMT) may be a critical mechanism underlying resistance. EMT is associated with increased invasiveness, motility, and a mesenchymal phenotype, which are hallmark features of chemoresistant cells [14]. This morphological transformation aligns with studies that link EMT to a more aggressive cancer phenotype and reduced chemosensitivity due to alterations in cell adhesion, increased motility, and enhanced survival pathways [15]. Notably, resistance developed at different cisplatin concentrations across cell line: MCF-7required higher cisplatin concentrations (5 µM) for resistance compared to MDA-MB-231 cells (1 µM), suggesting variations in the activation of resistance pathways. This divergence may reflect differences in baseline sensitivity and cellular adaptation, with triple-negative cancer cells like MDA-MB-231 potentially harbouring inherent mechanisms favouring rapid resistance Baykara, et al.
Cisplatin as a Suppressor of Cell Migration and Metastatic Potential
Cisplatin’s significant reduction in the migration ability of MCF-7 and MDA-MB-231 cells, as shown in the wound-healing assays, suggests an added benefit of this drug in potentially inhibiting metastatic spread. In untreated controls, the MDA-MB-231 cells exhibited rapid wound closure, a hallmark of their aggressive and highly migratory nature [17]. However, with cisplatin treatment (100 µM for MDA-MB-231), migration was substantially reduced, indicating that the drug effectively impairs TNBC cell motility at higher concentrations. This aligns with the notion that, beyond direct cytotoxicity, cisplatin may impede the metastatic cascade by reducing cell migration, thereby potentially limiting secondary tumor formation. The inhibition of migration, particularly in the MDA-MB-231 cell line, may also be indicative of cisplatin’s impact on signalling pathways associated with motility and EMT. Specifically, cisplatin is known to interfere with pathways involving matrix metalloproteinases (MMPs), which play a key role in cell migration and invasion [16]. Thus, these results suggest that cisplatin’s anti-metastatic effects in highly aggressive breast cancer types could further enhance its clinical value when used in combination with other targeted therapies aimed at reducing EMT and metastatic behaviour. This research demonstrates that cisplatin both cytotoxic and anti-migratory activity against breast cancer cell lines can be of major importance for treating TNBC. The increased sensitivity associated with longer cisplatin therapy points to the need for optimising doses to maximise efficacy. But the development of resistant phenotypes presents a big problem, so combinations therapies for resistance mechanisms must be developed. The morphological changes of resistance tell us that EMT is the heart of cisplatin tolerance. The EMT was previously associated with increased invasiveness, mobility and chemoresistance, so it can be a therapeutic target [4, 18]. This migration decrease also means that cisplatin might also block metastatic potential, offering a therapy extra to its cytotoxic nature. The anti-migratory response is probably due to interference with signalling pathways controlling motility, like matrix metalloproteinases (MMPs) [19]. Implications and Future Directions This study highlights the potential of cisplatin as a multi- functional chemotherapeutic agent that not only reduces cell viability but also inhibits cell migration, offering a dual approach to combating TNBC. However, the emergence of resistance remains a significant barrier, as illustrated by the establishment of cisplatin-resistant cell lines. Targeting the pathways involved in resistance development, such as EMT, RAN overexpression, or drug efflux pumps, could enhance cisplatin’s efficacy and reduce the likelihood of resistance. This approach may involve co-targeting strategies using inhibitors of EMT-related pathways or agents that block RAN GTPase function, which is implicated in chemoresistance in aggressive cancers [9]. Further research should explore the molecular basis of the observed resistance, focusing on identifying specific EMT markers, RAN GTPase activity, and drug transporters upregulated in resistant cells. Such studies could offer valuable insights into overcoming resistance and improving cisplatin’s therapeutic efficacy, especially for TNBC patients who currently have limited treatment options. Furthermore, research on the molecular mechanism of cisplatin resistance and on how to use co-targeting to make it more effective should be explored. Pairing cisplatin with agents that inhibit EMT or RAN could be a potential way to beat resistance and optimize outcomes in TNBC patients.
In conclusion, this study supports the use of cisplatin in breast cancer treatment, particularly for TNBC, where it not only reduces cell viability but also inhibits migration, potentially reducing metastasis. However, the risk of resistance emphasises the need for novel therapeutic strategies targeting resistance pathways to fully harness cisplatin’s clinical benefits in treating aggressive cancer types.
Clinical Application for this Research
The study shows that cisplatin’s efficacy increases over longer exposure times, clinically, this features the potential benefit of sustained, carefully timed cisplatin administration to maximise cancer cell cytotoxicity. The study provides critical insights that can help refine cisplatin’s clinical applications, particularly in addressing the challenges of TNBC treatment. Moreover, the emergence of cisplatin- resistant phenotypes, linked to epithelial-mesenchymal transition (EMT), underlines the need for co-targeting resistance pathways. Clinically, combining cisplatin with agents that inhibit EMT markers or associated pathways (e.g., matrix metalloproteinases or RAN GTPase) could decrease resistance and improve outcomes in aggressive cancers like TNBC. Also, the development of Tailored Therapies may be considered as from this study the differential findings for MCF-7 (ER-positive) and MDA-MB-231 (TNBC) cells can guide the personalisation of cisplatin-based therapies. Covering cell-specific responses and resistance mechanisms permits clinicians to modify treatments, such as combining cisplatin with hormone therapy for ER-positive cases or immune-modulating agents for TNBC.
In conclusion, this study supports the use of cisplatin in breast cancer treatment, particularly for TNBC, where it not only reduces cell viability but also inhibits migration, potentially reducing metastasis. However, the risk of resistance emphasises the need for novel therapeutic strategies targeting resistance pathways to fully harness cisplatin’s clinical benefits in treating aggressive cancer types.
Acknowledgement
We would like to acknowledge the institute of cancer therapeutics for funding the research.
Conflict of Interest
There is no conflict of interest in anyway in this research between authors.
References
-
Sung H, Ferlay J, Siegel RL, Laversanne M, Jemal A, et al. (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 71(3): 209-249.
-
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, et al. (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 68(6): 394-424.
-
Zong Y, Pegram M (2021) Research advances and new challenges in overcoming triple-negative breast cancer. Cancer Drug Resistance 4(3): 517-542.
-
Ebrahimi N, Manavi MS, Faghihkhorasani F, Fakhr SS, Baei FJ, et al. (2024) Harnessing function of EMT in cancer drug resistance: a metastasis regulator determines chemotherapy response. Cancer and Metastasis Reviews 43(1): 457-479.
-
Obidiro O, Battogtokh G, Akala EO (2023) Triple negative breast cancer treatment options and limitations: future outlook. Pharmaceutics 15(7): 1796.
-
Elrewey HA (2020) The effect of RAN inhibition on human colorectal cancer cells (CRC). University of Bradford.
-
Kulkarni B, Kirave P, Gondaliya P, Jash K, Jain A, et al. (2019) Exosomal miRNA in chemoresistance, immune evasion, metastasis and progression of cancer. Drug Discovery Today 24(10): 2058-2067.
-
Mohammadi A (2023) RAN GTPase in breast cancer: A key player in metastasis and chemoresistance. Cell Rep Med 4(3): 100871.
-
Kurisetty V (2008) RAN GTPase is a novel prognostic marker in epithelial ovarian cancer and is overexpressed in aggressive breast cancers. J Clinical Invest 118(4): 1555-1568.
-
Yuen HF (2012) Ran GTPase is a potential marker for cancer progression. J Clin Pathol 65(3): 236-242.
-
Şerban C, Anca M, Raica M (2015) The Story of MCF- 7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res 35(6): 3147-3154.
-
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, et al. (2012) Molecular mechanisms of cisplatin resistance. Oncogene 31(16): 1869-1883.
-
Carey L, Winer E, Viale G, Cameron D, Gianni L (2010) Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 7(12): 683-692.
-
Thiery JP, Huang RY, Nieto MA, Huang RY (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139(5): 871-890.
-
Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29(34): 4741-4751.
-
Fu S (2015) Effect of cisplatin on MMP expression in breast cancer cells. J Oncol.
-
Shafee N, Smith CR, Wei S, Kim Y, Mills GB, et al. (2008) Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res 68(9): 3243-3250.
-
Zhang Y (2023) EMT and cancer progression: Targeting mesenchymal markers to overcome drug resistance. Cancer Res 83(10): 1782-1795.
-
Chatterjee N, Walker GC (2023) Mechanisms of cisplatin resistance in cancer: DNA damage and beyond. Trends in Cancer 9(1): 15-29.
-
Alonso CD (2023) Targeting cell migration in triple- negative breast cancer. Sci Adv 9(5): 2789.
-
Duffy MJ (2023) Emerging biomarkers in TNBC: Novel strategies for drug resistance and metastasis. Nature Cancer 4(2): 120-134.
-
Ren Y (2012) Role of RAN in cancer progression and therapy resistance. Cancer Letters 324(2): 123-131.
-
Rottenberg S, Jonkers J (2012) Tackling the problem of resistance in triple-negative breast cancer. Drug Resistance Updates 15(2): 103-110.
-
Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, et al. (2002) Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346(2): 92-98.
-
Siegel RL, Miller KD, Wagle NS, Jemal A (2023) Cancer statistics, 2023. CA Cancer J Clin 73(1): 17-48.
- Pattern of Gonadal Hormones in Oral Testosterone-Supplimented Male Wistar Rats with Diabetes-Induced Hypogonadism
- Re-Evaluation of the Genotoxicity of Currently Used Food Dyes in Mouse Multiple Organs Via Continuous Administration by Drinking Using the Comet Assay
- Pharmacogenetics of Type 2 Diabetes Mellitus: Linking Genetic Variability to Drug Efficacy and its Cardiovascular Outcomes
- Exploratory Proteomic Profiling of SARS-CoV-2 Infected THP-1 Macrophages Reveals Alterations in Inflammatory Response and Cellular Metabolism
- Study of Genotoxicity of Hepatocarcinogens in Multiple Organs in Mice by Feeding and Drinking Using the Comet Assay
- Spirulina Polypeptides Inhibit the Growth of Human Lung Tumor (H460) Cells