Bioequivalence of Oseltamivir Phosphate 6 mg/mL Powder for Oral Suspension Formulations in Healthy Thai Volunteers under Fasting Conditions
Oseltamivir is an oral antiviral of choice indicated for treatment and prophylaxis of influenza A and B infections. Oseltamivir is an inactive prodrug of oseltamivir carboxylate, an active form exerting antiviral activity. Oseltamivir oral suspension is an alternative formulation for pediatric patients or adults with dysphagia. GPO-A-FLU®, oseltamivir 6 mg/mL powder for oral suspension had been developed as a generic substitute for reference product, Tamiflu® oral suspension. In this study, we investigated the pharmacokinetics and bioequivalence of the test oral suspension oseltamivir formulation with respect to the corresponding reference oral suspension formulation. A comparative open-label, randomized, single dose, two-way crossover study was conducted under fasting conditions. Blood samples were collected for 24 hours post-dose and the plasma was separated for oseltamivir assay using a validated liquid chromatography-mass spectrometry method. The pharmacokinetic parameters were determined from plasma concentration-time profiles after administration of the test and reference formulations. The pharmacokinetic parameters were in agreement with the previously published data. The primary pharmacokinetic parameters: AUC0−tlast, AUC0− and Cmax obtained from 47 subjects who completed the study were statistically compared. The 90% confidence intervals of geometric least squares mean ratio (test/reference) for log-transformed parameters were within 80.00-125.00% of bioequivalence criteria: 92.33-98.52% for AUC0−tlast, 92.28- 98.43% for AUC0−, and 81.82-94.26% for Cmax. Both products were generally well tolerated by healthy Thai subjects. This study successfully demonstrated bioequivalence between GPO-A-FLU® and Tamiflu® and supported product registration.
Introduction
Oseltamivir is an oral antiviral of choice indicated for treatment and prophylaxis of influenza A and B infections [1, 2, 3]. Oseltamivir is an inactive prodrug and needs to be hydrolyzed by esterase enzyme to active form, oseltamivir carboxylate to exert antiviral activity. This active metabolite binds to neuraminidase which enables release of virions from the infected cells and facilitates viral replication [2, 3, 4]. A standard treatment dose of adult is 75 mg twice-daily for 5 consecutive days. It is recommended to start the treatment as early as possible, preferably within 48 hours of symptoms. For prophylaxis, once-daily standard dose within 48 hours of expose is recommended. The dosage of pediatric patient is adjusted by body weight [4, 5].
After oral administration, oseltamivir is readily absorbed from the gastrointestinal tract with 80% absolute bioavailability. There is no food effect on the absorption. Protein binding of oseltamivir is approximately 42% which is higher than the binding of its active form. Oseltamivir is primarily and rapidly converted by hepatic carboxylesterase and subsequently eliminated by renal excretion. Thus, dose adjustment is recommended for patients with renal impairment. Apparent elimination half-life of oseltamivir ranges between 1 and 3 hours whereas longer half-life has been observed for oseltamivir carboxylate [1, 2, 3, 4].
Oseltamivir oral suspension is an alternative formulation for pediatric patients or adults with dysphagia. GPO-A-FLU®, oseltamivir 6 mg/mL powder for oral suspension had been developed as a generic substitute for reference product, Tamiflu® oral suspension. The objective of this study was to determine the pharmacokinetic parameters of the test and reference oseltamivir oral suspension formulations and then to statistically compare the parameters to evaluate bioequivalence between these formulations.
Materials and Methods
Study Products
GPO-A-FLU®, oseltamivir 6 mg/mL powder for oral suspension (Lot No. S620085) manufactured by the Government Pharmaceutical Organization (GPO), Thailand was used as the test formulation and Tamiflu® (Lot No. B8061) manufactured by Rottendorf Pharma GmbH, Germany was used as the reference formulation.
Chemicals and Reagents
Oseltamivir (Figure 1A) working standard bearing Lot No. 9-TMH-42-2, 96% purity was purchased from Toronto Research Chemicals (Toronto, Canada). Oseltamivir D5 hydrochloride (Figure 1B) working standard bearing Lot No. CS-OE-391, 91.69% purity was purchased from Clearsynth Labs Ltd. (Mumbai, India).
![Figure 1: Chemical structure of oseltamivir [A] and oseltamivir D5 hydrochloride [B]](/fulltextimages/6778/fig_1.png)
Ultrapure water was produced in-house using a GenPure PRO UV-TOC Ultrapure Water System (Thermo Fisher Scientific; USA). All solvents were HPLC grade. Chemicals and reagents were analytical grade.
Study Subjects
The number of subjects was determined by considering the estimated intra-subject variability about 27% [6], T/R ratio 95%, significance level 5%, power ≥ 90% and bioequivalence limits of 80.00-125.00%. Based on the calculation, the sample size of 42 subjects would be sufficient to demonstrate bioequivalence. However, a total of 52 subjects were enrolled in this study by estimating 20% dropout rate. The study subjects were healthy males and females at the age between 18 and 55 years with a body mass index (BMI) between 18 and 30 kg/m2. They had no evidence of underlying disease or clinically significant abnormal laboratory findings. Female subjects were not pregnant or breastfeeding at all time of the study. Females with childbearing potential were instructed to use acceptable methods of birth control including non- hormonal methods and sexual abstinence throughout the entire of the study.
The subjects were not enrolled or withdrawn from the study according to the exclusion criteria including history of hypersensitivity to oseltamivir or any components of the product, allergic reaction after taking any medications, alcohol dependence, drug abuse, cigarette smoking, recent blood donation, and recent clinical trial participation. Consumption of xanthine containing products, grapefruit/ pomelo/ orange-based products, or any medications was restricted prior to dosing and during entire study. All study subjects were well informed and provided the written informed consent before study participation at International Bio Service Co., Ltd., Golden Jubilee Medical Center, Mahidol University, Thailand.
Study Design
The bioequivalence study of two oseltamivir 6 mg/mL powder for oral suspension formulations was designed as a comparative open-label, randomized, single dose, two- way crossover study. Fifty-two study subjects were enrolled in the study and randomly divided to two groups, test- reference (TR) and reference-test (RT) meaning that the study subjects received either the test or reference product in period I and switched over to another product in period II after 7-day washout period. After at least 10-hour overnight fasting, each subject received 12.5 mL (equivalent to 75 mg of oseltamivir) of the assigned formulation with 240 mL of water. The clinical study protocol was reviewed and approved by the Institute for the Development of Human Research Protections, Thailand on 9 January 2020 (COA
No. IHRP2020005). The study was conducted strictly in accordance with ICH Guidance on Good Clinical Practice and Declaration of Helsinki. Health and wellbeing of the subjects were monitored throughout the study by direct questioning, clinical examination, and laboratory testing.
Blood Sampling
Twenty-four blood samples were collected from each subject in each period at 0 (pre-dose), 0.17, 0.33, 0.5, 0.67, 0.83, 1, 1.25, 1.5, 1.75, 2, 2.25, 2.5, 2.75, 3, 3.5, 4, 5, 6, 8, 10, 12, 16 and 24 hours post-dose. Approximate 4 mL of each blood sample was drawn from an indwelling cannula in the forearm vein using syringe and transferred into vacutainers containing sodium fluoride 15 mg and potassium oxalate 12 mg as anticoagulant (BD Hemogard™, USA). The vacutainers were gently inverted several times to ensure the mixing of tube contents and placed upright in wet ice water bath maintained below 5°C until centrifugation. Plasma sample was achieved from centrifugation at 3000±100 relative centrifugal force (rcf) for 5 minutes at 4°C. Plasma separation was done within 30 minutes from the blood collection. The separated plasma samples were divided and transferred to polypropylene tubes in two separate aliquots and stored upright in the freezer maintained at -50° or colder until analysis.
Sample Analysis and Incurred Sample Reanalysis (ISR)
The plasma samples were analyzed at GPO, Thailand complying with the Principles of Good Laboratory Practice, European Medicines Agency (EMA) guideline on the investigation of bioequivalence [7] and in-house SOPs. The samples were analyzed along with 8 calibration standards covering a range of concentration between 0.105 and 164.916 ng/mL, and 4 sets of quality control samples at 4 concentration levels. Working solution of internal standard (IS), 20 ng/mL of oseltamivir D5 at 50 µL was added into 250 µL of samples. Thereafter, 3 mL of extraction solvent (tert- butyl methyl ether-dichloromethane-hexane (7:2:1, v/v/v) was added. The samples were centrifuged at 3500 rcf for 5 minutes at 10°C. Then the plasma layer was flash-frozen, and the organic layer was transferred into pre-labeled tube. The contents were evaporated at 40°C to dryness, then reconstituted with 250 µL of methanol-acetonitrile-0.05% v/v formic acid (3:3:2, v/v/v). The processed samples were transferred into HPLC vials for subsequent analysis.
Sample analysis was performed on a Shimadzu Nexera UPLC system (Shimadzu Corporation, Japan). The chromatographic separation was achieved using Symmetry® C18 (3.9×150 mm, 5 µm) analytical column maintained at 50°C. An isocratic mobile phase consisting of methanol- acetonitrile-0.05% v/v formic acid (22.5:22.5:55, v/v/v) was delivered into the system at a flow rate of 0.7 mL/min. The samples were kept in autosampler at 4°C and injected at 10 µL. The detection was done using electrospray ionization triple quadrupole mass spectrometry (TSQ Quantum Ultra, Thermo Fisher Scientific Inc., USA). The multiple reaction monitoring (MRM) was carried out in positive mode with precursor/product transitions of m/z 313.038 to 166.080 and m/z 318.058 to 171.110 for oseltamivir and IS, respectively. Data analysis was performed using XcaliburTM 4.0.27.42 and LCquanTM 3.0.26.0 (Thermo Fisher Scientific Inc., USA).
Incurred sample reanalysis was performed to ensure the reproducibility and reliability of the study data. According to EMA guideline on bioanalytical method validation [8], study samples having concentrations close to Cmax and in the elimination phase of each subject in each period were chosen and reanalyzed. At least 10% of first 1000 samples and 5% of the number of samples exceeding 1000 samples were reanalyzed. The concentration values from incurred samples were not used for pharmacokinetic calculation.
Pharmacokinetic and Statistical Analysis
The pharmacokinetic parameters were calculated using non-compartmental model of Phoenix WinNonlin Professional software version 6.4 (Pharsight Corporation, USA). The maximum concentration (Cmax) and time to achieve maximum concentration (tmax) were directly obtained from the concentration-time data. Area under the concentration-time curve from time zero to the last measurable concentration (AUC0-tlast), area under the concentration-time curve from time zero to infinity (AUC0-∞) and Cmax were the primary pharmacokinetic parameters used for bioequivalence evaluation while tmax, elimination rate constant (λ2) and half- life (t1/2) were considered as the secondary parameters.
The statistical analysis was performed using PROC GLM of SAS® version 9.4 (SAS Institute Inc., USA). Analysis of variance (ANOVA) was carried out for log-transformed of primary parameters (AUC0-tlast, AUC0-∞ and Cmax). ANOVA model included sequence, formulation and period as fixed effects. However, subject nested within sequence was included as a random effect, as well as an error term for sequence effect testing. The statistical significance of the effects involved in the model was determined using F-test. Bioequivalence of two formulations was concluded, if the 90% confidence intervals (CIs) for the geometric least square mean ratios (test/reference) of log-transformed primary pharmacokinetic parameters were within the acceptance range of 80.00-125.00%. Wilcoxon signed-rank test was performed to compare the median tmax of the test and reference formulations. All statistical calculations were performed at a significance level of 5% (α=0.05). Results
Demographic Characteristic of Subjects
Fifty-two adult Thai male and female volunteers were enrolled and randomly divided into TR and RT group equally. There were 13 males and 13 females in each group. The mean ± SD of age, height, weight and BMI were 35.87±8.8 years, 1.62±0.08 m, 61.13±9.45 kg and 23.07±2.61 kg/m2, respectively. There were total five dropout and withdrawn subjects. Three subjects were withdrawn from the study due to adverse event and safety concern. Two subjects dropped out before check-in to period II due to personal reasons. Therefore, 47 subjects completed the study and their plasma concentration data were used for pharmacokinetic and statistical analysis.
Study Sample Analysis and Incurred sample Reanalysis (ISR)
All study samples from 52 subjects including withdrawn and dropout subjects were completely analyzed. The samples from the same subject were analyzed in the same analytical run. The correlation coefficient calculated from 8 calibration standards was more than 0.99 for all analytical runs. The precision and accuracy of the analysis were demonstrated using quality control samples in each analytical run. The coefficient of variation (CV) and accuracy of quality control samples in each analytical run ranged from 2.3% to 6.5% and 98.2% to 100.6%, respectively. The study samples analysis was completed within 39 days since collecting blood samples, which were within the established long-term stability in method validation.
Incurred sample reanalysis was carried out to prove the performance of bioanalytical method in analyzing subject samples. As per regulatory guideline [8], at least 169 samples should be selected from total 2,372 subject samples. A total of 198 samples were chosen for reanalysis and 100% of reanalyzed samples showed that the percent difference between the original and reanalyzed concentration was less than 20%. The results indicated good reproducibility and reliability of the analytical method.
Pharmacokinetic and Statistical Analysis
Plasma concentrations of oseltamivir were analyzed as a function of time for both the test and reference formulations (Figure 2).
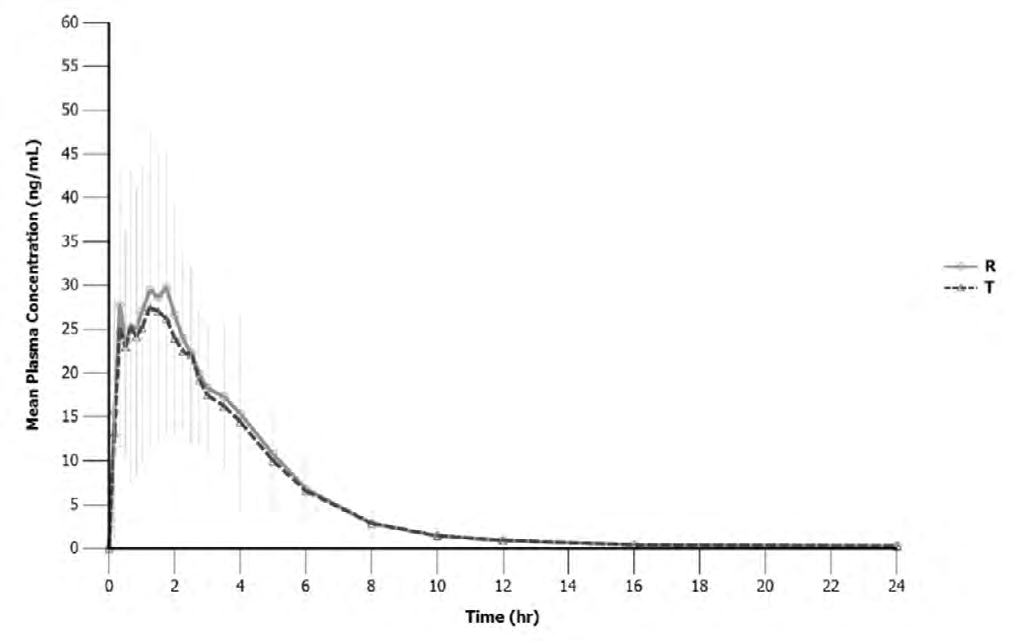
The mean ± SD values of the pharmacokinetic parameters for two formulations from 47 subjects were represented in Table 1.
| Parameters | Mean ± SD | |
|---|---|---|
| Test | Reference | |
| AUC (ng.hr/mL) 0-tlast | 126.2 ± 36.1 | 133.2 ± 43.3 |
| AUC (ng.hr/mL) 0-∞ | 127.9 ± 36.3 | 134.9 ± 42.9 |
| C (ng/mL) max | 37.1 ± 17.2 | 42.0 ± 18.2 |
| t (hr)a) max | 1.25 (0.17, 4.00) | 1.50 (0.17, 4.00) |
| λ (1/hr) z | 0.23 ± 0.08 | 0.22 ± 0.07 |
| t (hr) 1/2 | 3.30 ± 1.19 | 3.72 ± 1.89 |
| Extrapolated AUC (%) | 1.42 ± 1.46 | 1.49 ± 1.58 |
Table 1: Pharmacokinetic parameters of test and reference formulations a)tmax is represented in median (min,max).
According to the results, the mean Cmax values of both formulations were comparable, 37.1 ng/mL for the test and 42.0 ng/mL for the reference. Oseltamivir was well absorbed after oral administration with tmax ranging between 0.17 and 4 hours for the test and reference.
Statistical analysis was carried out on the data obtained from the subjects who completed the study (N=47). The results of ANOVA showed insignificant effect of sequence and period on log-transformed AUC0-tlast, AUC0-∞ and Cmax (p>0.05), but significant formulation effect was observed on all tested parameters. The 90% CIs for the geometric least square mean ratios of log-transformed AUC0-tlast, AUC0-∞ and Cmax between the two formulations were within the acceptance range of 80.00-125.00% as summarized in Table 2.
| Parameters | Ratio (90% CI) | Power | Intra-subject CV (%) | ANOVA (p-value) | ||
|---|---|---|---|---|---|---|
| ln AUC 0-tlast | 95.4 (92.33 - 98.52) | 100% | 9.4 | 0.5124 | 0.0181 | 0.2893 |
| ln AUC 0-∞ | 95.3 (92.28 - 98.43) | 100% | 9.3 | 0.4872 | 0.0160 | 0.3044 |
| ln C max | 87.8 (81.82 - 94.26) | 100% | 20.6 | 0.1830 | 0.0035 | 0.7996 |
Table 2: Statistical comparison of pharmacokinetic parameters between test and reference formulation.
Wilcoxon signed-rank test was performed and indicated no significant difference in median tmax between the test and reference formulations (p>0.05).
Tolerability
Concerning to the tolerability and welfare of study subjects, adverse events were monitored closely and recorded throughout the study. A total of 17 post-dose adverse events were reported in 12 study subjects as represented in Table 3.
| Adverse event | Reported adverse event incidence (N) | |
|---|---|---|
| Test product | Reference product | |
| Nausea | 5 | 5 |
| Diarrhea | 0 | 1 |
| Vomiting | 1 | 1 |
| Dry mouth | 0 | 1 |
| Fever | 0 | 1 |
| Dizziness | 1 | 1 |
| Total adverse events | 7 | 10 |
Table 3: List of adverse events.
Seven adverse events were reported in 6 subjects after receiving the test formulation whereas 10 adverse events were reported in 8 subjects after receiving reference formulation. The most frequently reported adverse event for both test and reference was nausea. All adverse events were mild in severity and could recover without any medical treatment. In addition, all adverse events were reported to the Institute for the Development of Human Research Protections, Thailand in a timely manner.
Discussion
A total of 52 subjects were enrolled in the study and five subjects did not complete the study. Two of them were withdrawn due to vomiting within 3 hours after dosing. The data from these subjects were excluded from statistical analysis because the vomiting occurred before 2 times of median tmax [9]. Based on the subject number calculation, the data from 47 subjects were sufficient for establishing bioequivalence with adequate power. The sampling time points were assigned more frequently during first 3 hours after dosing in order to capture Cmax in the absorption phase and avoid Cmax being the first time-point of a pharmacokinetic profile. Moreover, the blood samples were collected until the last time point at 24 hours post-dose to ensure that the sampling period would cover at least 3 elimination half-lives [9].
Although a prodrug, oseltamivir is primarily and rapidly converted by hepatic carboxylesterase after oral administration, it can also be converted by esterase enzymes in blood and plasma [10]. The use of sodium fluoride, a general esterase inhibitor is well justified in preventing ex-vivo conversion of oseltamivir in blood or plasma sample [10, 11, 12, 13]. In addition, a commercially available sodium fluoride collection tube is convenient for use in the bioequivalence study as many samples need to be collected. In-house stability data showed that using sodium fluoride and potassium oxalate as anticoagulant could maintain the integrity of the study samples for oseltamivir assay. Therefore, a commercial vacutainer containing sodium fluoride and potassium oxalate were used for blood collection throughout the study. However, for stability purposes, blood samples were controlled in wet ice water bath maintained below 5°C immediately after collection until centrifugation, and plasma separation was done strictly within 30 minutes.
Plasma concentration of oseltamivir in the study samples were determined by a liquid chromatography tandem mass spectrometry method which was validated following the Guideline on bioanalytical method validation of European Medicines Agency (EMA) [8] and the U.S.FDA Guidance for Industry Bioanalytical Method Validation [14]. Even though oseltamivir is inactive, demonstration of bioequivalence by comparing pharmacokinetics of parent drug is recommended rather than its metabolite because the absorption of parent drug is more relevant and sensitive to detect the differences between the test and reference formulations [6, 7, 15].
The pharmacokinetic parameters and profiles were
comparable between the test and reference formulations. The pharmacokinetics of oseltamivir characterized in this study corresponded to the results of previous study in Thai population receiving oseltamivir capsules under fasting condition [16]. Moreover, the pharmacokinetic data were in agreement with the data in different ethnicities [17, 18, 19]. The intra-subject variability of log-transformed AUC0-tlast, AUC0-∞ and Cmax obtained in this study were less than 30% of the CV indicated that oseltamivir was not highly variable drug. The ANOVA model showed a significant formulation effect on all primary pharmacokinetic parameters (p<0.05). These might be because the intra-subject variability of each parameter was lower than expected and the number of subjects providing data for statistical comparison was high. However, the bioequivalence was concluded by 90% CIs which were within the acceptance criteria of 80.00-125.00%. Thus, a significant formulation effect did not interfere in the results of this study [20].
Oseltamivir was generally well tolerated and no serious drug-related adverse events were reported [1, 2, 3, 4]. Only mild adverse events e.g. nausea, diarrhea, vomiting, dry mouth, fever and dizziness were observed in this study. The incidence of adverse events reported after receiving the test and reference formulation was comparable.
Conclusions
A comparative open-label, randomized, single dose, two-way crossover study successfully demonstrated bioequivalence between GPO-A-FLU® and Tamiflu® oseltamivir (6 mg/mL) oral suspension. Both formulations were generally well tolerated by healthy Thai subjects and no serious adverse events were reported. The data presented in the study met the regulatory criteria and supported product registration.
Acknowledgements
We are thankful to the Government Pharmaceutical Organization for supporting the study. We would also like to acknowledge the encouragement of our colleagues and the collaboration from the clinical team at International Bio Service Co., Ltd.
References
-
Bardsley Elliot A, Noble S (1999) Oseltamivir. Drugs 58(5): 851-860.
-
McClellan K, Perry CM (2001) Oseltamivir: a review of its use in influenza. Drugs 61(2): 263-283.
-
He G, Massarella J, Ward P (1999) Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin Pharmacokinet 37(6): 471-484.
-
Davies BE (2010) Pharmacokinetics of oseltamivir: an oral antiviral for the treatment and prophylaxis of influenza in diverse populations. J Antimicrob Chemother 65 Suppl 2(2): ii5–ii10.
-
Uyeki TM, Bernstein HH, Bradley JS, Englund JS, File TM, et al. (2019) Clinical Practice Guidelines by the Infectious Diseases Society of America: 2018 Update on Diagnosis, Treatment, Chemoprophylaxis, and Institutional Outbreak Management of Seasonal Influenzaa. Clin Infect Dis an Off Publ Infect Dis Soc Am 68(6): 895-902.
-
WHO (2019) Notes on the Design of Bioequivalence Study : Oseltamivir. WHO Prequalification Team: Medicines, Geneva, World Health Organization, Switzerland, pp: 2.
-
EMA (2010) Guideline on the Investigation of Bioequivalence. Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency, London.
-
EMA (2011) Guideline on bioanalytical method validation. Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency, London, pp:1- 23.
-
FDA (2013) Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA. Center for Drug Evaluation and Research (CDER), Silver Spring, Food and Drug Administration, U.S.
-
Lindegardh N, Davies GR, Tran TH, Farrar J, Singhasivanon P, et al. (2006) Rapid degradation of oseltamivir phosphate in clinical samples by plasma esterases. Antimicrob Agents Chemother 50(9): 3197-3199.
-
Kromdijk W, Rosing H, van den Broek MPH, Beijnen JH, Huitema ADR (2012) Quantitative determination of oseltamivir and oseltamivir carboxylate in human fluoride EDTA plasma including the ex vivo stability using high-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J Chromatogr B, Anal Technol Biomed life Sci 891-892: 57-63.
-
Lindegårdh N, Hanpithakpong W, Wattanagoon Y, Singhasivanon P, White NJ, et al. (2007) Development and validation of a liquid chromatographic-tandem mass spectrometric method for determination of oseltamivir and its metabolite oseltamivir carboxylate in plasma, saliva and urine. J Chromatogr B, Anal Technol Biomed life Sci 859(1): 74-83.
-
Hu ZY, Laizure SC, Meibohm B, Herring VL, Parker RB (2013) Simple and sensitive assay for quantification of oseltamivir and its active metabolite oseltamivir carboxylate in human plasma using high-performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry: Improved applicabili. J Pharm Biomed Anal 72: 245-250.
-
FDA (2018) Guidance for industry: Bioanalytical method validation. Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM), Silver Spring, Food and Drug Administration, U.S, pp: 1-44.
-
European Medicines Agency (2015) Oseltamivir hard capsules 30 , 45 and 75 mg , powder for oral suspension 6 mg / ml and 12 mg / ml product-specific bioequivalence guidance *. Committee for Medicinal Products for Human Use (CHMP), London, pp: 2.
-
Kongpatanakul S, Chatsiricharoenkul S, Panich U, Sathirakul K, Pongnarin P, et al. (2008) A randomized, open-label, 2-period, crossover bioequivalence study of two oral formulations of 75 mg oseltamivir in healthy Thai volunteers. Int J Clin Pharmacol Ther 46(12): 654- 662.
-
Yun YL, Gao SH, Wen Y, Wang ZP, Miao HJ, et al. (2017) Bioequivalence of two oseltamivir formulations in healthy Chinese volunteers . Int J Clin Pharmacol Ther 55(9): 761-768.
-
Schentag JJ, Hill G, Chu T, Rayner CR (2007) Similarity in pharmacokinetics of oseltamivir and oseltamivir carboxylate in Japanese and Caucasian subjects. J Clin Pharmacol 47(6): 689-696.
-
Medina Nolasco A, Ortiz Campos K, López Bojórquez E, Zazueta Beltrán L, et al. (2018) Bioequivalence of Two Oral-Suspension Formulations of Oseltamivir in Healthy Mexican Adults. Am J Pharmacol Ther 2(1): 7-11.
-
Bapuji A, Ravikiran HLV, Nagesh M, Syedba S, Ramaraju D, et al. (2010) Bioequivalence Testing - Industry Perspective. J Bioequiv Availab 2(5): 98-101.
- Effects of 5-HTP and Melatonin on the Sleep Cycle of Medical Students
- Adsorption of Bisphenol A on NH4OH- Modified Rice Husk and Sugar Cane Bagasse Biochar
- Comparative Assessment of the Reinforcement Efficiency of Palm Fruit Fibre and Coconut Fibre in High Density Polyethylene (HDPE) Matrix Composite
- Importance of Bio Compounds Naturally Present in Food with Functionality in Animal Metabolism
- Sub-Acute Study on the Cardiotoxic Effects of Monosodium Glutamate Ingestion in Albino Rat
- Weight Management and Its Natural Solutions: A Review